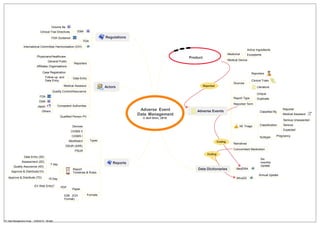
This document discusses guidelines and regulations for clinical trials and adverse event reporting from various international organizations such as the International Committee on Harmonisation (ICH), FDA, and EMA. It outlines the actors and roles involved in the processes of case registration, data entry, assessment, quality control, and reporting of data from clinical trials, physicians, and the general public on adverse events related to medical products and devices. The diagram also shows the various formats, timelines, sources, and classifications used to manage this post-marketing safety data.