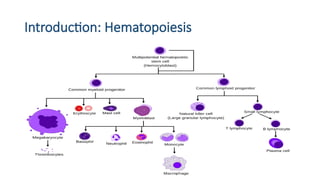
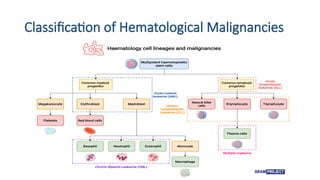
Introduction
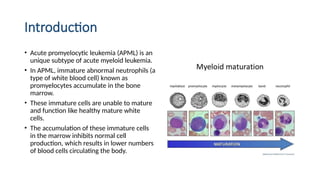
• Acute promyelocyticleukemia (APML) is an
unique subtype of acute myeloid leukemia.
• In APML, immature abnormal neutrophils (a
type of white blood cell) known as
promyelocytes accumulate in the bone
marrow.
• These immature cells are unable to mature
and function like healthy mature white
cells.
• The accumulation of these immature cells
in the marrow inhibits normal cell
production, which results in lower numbers
of blood cells circulating the body.
6.
APML
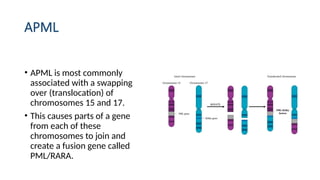
• APML ismost commonly
associated with a swapping
over (translocation) of
chromosomes 15 and 17.
• This causes parts of a gene
from each of these
chromosomes to join and
create a fusion gene called
PML/RARA.
7.
History
APML was firstdescribed in 1957 by a Swedish
author, Hillestad, when he reported 3 patients
characterized by “A very rapid fatal course of only a
few weeks duration,” with a white blood cell (WBC)
picture dominated by promyelocytes and a severe
bleeding tendency.
• More detailed features of APML were described by
Bernard et al in 1959, and the severe hemorrhagic
diathesis has been described to disseminated
intravascular coagulation (DIC) or hyperfibrinolysis.
9.
Epidemiology
• Acute promyelocyticleukemia (APL), is a distinct
subtype of acute myeloid leukemia, represents
about 8% - 15% of pediatric APML.
• The disease occur at any age but patients are
predominantly adult or in midlife. Older child and
adolescents are the major risk group in case of
childhood APML.
10.
.
Laboratory evidenceof DIC is present in 70%
to 80% of patients at diagnosis or shortly after.
Hemorrhagic events contribute 10% to 15% excess
mortality during induction chemotherapy for APML.
Morphologically, it is identified as aml-m3 by the
French-American-British (FAB) classification.
11.
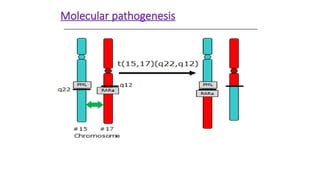
Cytogenetically ,APL is characterized by a
balanced reciprocal translocation abnormality,
t(15;17)(q22;q12); PML-RARA.
Currently it is one of the most treatable forms of
leukemia with a 12-yr PFS rate, is estimated to be
approximately 70%.
.
In Acutepromyelocytic leukemia (APL), there is an
abnormal accumulation of immature granulocytes
called promyelocytes.
APL is characterized a balanced reciprocal
translocation abnormality, t(15;17)(q22;q12); PML-
RARA, which results in fusion of the retinoic acid
receptor (RARA) gene on chromosome 17 with the
promyelocytic leukemia (PML) gene on
chromosome 15.
.
. The fusionof PML and RARA results in expression of
a hybrid protein with altered functions. This fusion
protein binds with enhanced affinity to sites on the
cell's DNA, blocking transcription and differentiation
of granulocytes.
Although the chromosomal translocation involving
RARA is believed to be the initiating event,
additional mutations are required for the
development of leukemia.
16.
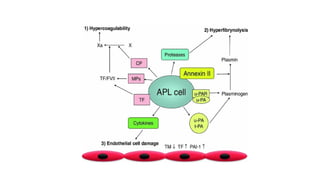
Fatal coagulopathy inAPML
APML is special from other leukemia because life threatening
bleeding is frequently associated with this type. Reasons are-
Procoagulant activity
APL cells express two tumor-associated procoagulants:
tissue factor (TF) and cancer procoagulant (CP).
Cytokines
Leukemic promyelocytes secrete various cytokines like
IL-1b and TNFa that may mediate APL-associated
coagulopathy through various complex mechanisms.
17.
.
Fibrinolysis
Leukemic promyelocyteshighly express
annexin-II, which may lead to primary fibrinolysis.
Annexin-II is a protein receptor with a strong affinity
for plasminogen and tPA, which results in greatly
increased conversion of plasminogen to plasmin.
Proteolysis
Proteolysis of clotting factors and fibrinogen by
granulocytic proteases such as elastase and
chymotrypsin, found in the granules of APML blasts.
19.
Presentation
• Most patientswith APML present with
pancytopenia. About 10-30% of patients
present with leukocytosis .
• APL differs from AML in that most patients
present with coagulopathy. The coagulopathy
has been described as disseminated
intravascular coagulation (DIC) with
associated hyperfibrinolysis.
• It is important to treat the coagulopathy as a
medical emergency. In 40% of untreated
patients, pulmonary and cerebral
hemorrhages can occur.
20.
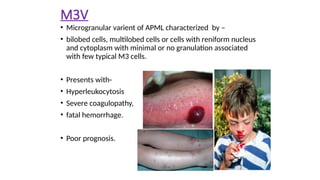
M3V
• Microgranular varientof APML characterized by –
• bilobed cells, multilobed cells or cells with reniform nucleus
and cytoplasm with minimal or no granulation associated
with few typical M3 cells.
• Presents with-
• Hyperleukocytosis
• Severe coagulopathy,
• fatal hemorrhage.
• Poor prognosis.
Treatment
Counseling
As APMLis a hematologic emergency, so immediate
treatment starting with ATRA when APML is
suspected regardless of cytogenetic/molecular study.
Supportive treatment
oHydration and prevention of TLS
oBlood product support
oOral care, Anal care, Antibiotics etc.
- It occursin 70% to 90% of cases.
- It occurs due to release of several procoagulants,
mainly tissue factor (TF), and cancer procoagulant
(CP).
-DIC complicated by APML is characterized by
exaggerated fibrinolysis and life-threatening
hemorrhage.
DIC in APML
30.
---Platelet transfusion tomaintain >50,000/cumm.
---FFP transfusion- To correct PT, APTT.
---Fibrinogen must be kept over 150 mg/dL.
Note:
Treatment
- Heparin is of no documented value.
- packed RBCs transfusion may worsen
the condition.
- Avoid invasive procedures if possible
(LP and central line placement).
31.
.
WBC Count RiskGroup
(10 X 109
/L or less ) Standard /Low risk
(>10 X 109
/L ) High risk
Risk stratification
32.
Induction Therapy –Low Risk APML
Preferred regimen (non-chemotherapy approach):
• - ATRA (Tretinoin): 45 mg/m²/day PO in 2 divided doses (until CR or max 60 days)
• - Arsenic trioxide (ATO): 0.15 mg/kg IV daily until marrow remission (max 60 doses)
Alternative:
• - ATRA + anthracycline-based chemotherapy:
• • Daunorubicin: 60 mg/m² IV days 1–3
• • Idarubicin: 12 mg/m² IV days 2, 4, 6, 8
• Notes: No cytarabine in low-risk disease. Monitor QTc, electrolytes, and signs of
differentiation syndrome.
33.
Induction Therapy –High Risk APML
Preferred regimen:
• - ATRA: 45 mg/m²/day PO (2 divided doses)
• - ATO: 0.15 mg/kg IV daily
Plus cytoreduction with one of the following:
• • Idarubicin: 12 mg/m² IV days 2, 4, 6, 8
• • Gemtuzumab ozogamicin: 9 mg/m² IV day 1
• • Daunorubicin: 60 mg/m² IV days 1–3
• • (Optional) Cytarabine: 100–200 mg/m²/day × 7 days
• Alternative (if anthracycline not tolerated):
• - ATRA + ATO + hydroxyurea 2–4 g/day PO until WBC <10 × 10⁹/L.
34.
Consolidation Phase
• Goal:Eliminate residual leukemia cells & reinforce remission.
• Low-risk APL:
• - ATRA: 45 mg/m²/day PO (2 weeks on, 2 weeks off) × 4 cycles
• - ATO: 0.15 mg/kg IV daily, 5 days/week × 4 weeks per cycle × 4 cycles
• High-risk APL:
• - Same as above ± idarubicin 5–12 mg/m² IV on day 1 per cycle
• - Optional: mitoxantrone 10 mg/m² IV or daunorubicin 50 mg/m² IV in selected protocols
• No maintenance if ATRA + ATO–only regimen and patient achieves molecular remission.
35.
Maintenance Therapy
• Goal:Maintain remission and prevent relapse (typically 1–2 years).
• Classic maintenance (for chemo-containing induction):
• - ATRA: 45 mg/m²/day PO × 15 days every 3 months
• - 6-Mercaptopurine: 50 mg/m² PO daily continuously
• - Methotrexate: 15 mg/m² PO weekly
• Omit maintenance if ATRA + ATO used alone and PCR for PML-RARA remains
negative.
36.
Relapsed/Refractory APL &Supportive Care
Re-treatment options:
• - ATRA + ATO if not previously combined
• - Add anthracycline if resistant
• - Stem cell transplant:
• • Autologous SCT for molecular remission
• • Allogeneic SCT for second relapse or persistent disease
• - Clinical trials for novel oral ATO or differentiation agents
• Supportive care:
• - Differentiation syndrome: Dexamethasone 10 mg IV q12h
• - Coagulopathy: Maintain platelets > 30–50 × 10⁹/L, fibrinogen >150 mg/dL
• - QT prolongation: Maintain K⁺ >4, Mg²⁺ >1.8
• - Infection prophylaxis and early febrile neutropenia management
37.
ATRA
• APML isunique among leukemias due to its
sensitivity to all-trans retinoic acid (ATRA; tretinoin),
the acid form of vitamin A.
• ATRA is fast acting and it can improve coagulopathy
within 48 hours to 5 days.
• Treatment with ATRA dissociates the repressor
complex from RAR and allows DNA transcription and
differentiation of the immature leukemic
• promyelocytes into mature granulocytes.
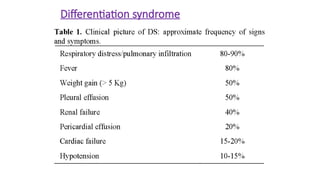
Differentiation syndrome
The differentiationsyndrome (previously called "retinoic acid
syndrome") is a potentially fatal complication of induction
chemotherapy in patients with acute promyelocytic leukemia
(APL). It usually occur in the first
1-2 weeks of treatment.
It is characterized by
• peripheral edema,
• Weight gain
• hypoxemia, respiratory distress
• hypotension,
• Unexplained fever
• renal and hepatic dysfunction,
• Rash and
• serositis resulting in pleural and pericardial effusion.
Treatment
• Temporary discontinuationof ATRA.
• Prompt initiation of Dexamethasone 10mg/m2 -12 hourly
until disappearance of signs and symptoms (minimum 3
days).
• Initiation of conventional Ara-C/daunorubicin
chemotherapy to control leukocytosis.
• Frusemide.
• Continue treatment with ATRA after controlling the
situation.
• Prophylaxis: Steroid commonly recommended,
in patients with elevated WBC.
43.
Arsenic Tri Oxide(ATO)
Despite its historical reputation as a toxin and poison,
ATO has been used in a variety of diseases for many
countries. ATO has now emerged as the treatment of
choice for patients with refractory/relapsed APML as
well as newly diagnosed APML.
Advantage:
- Early remission.
- Can cross blood brain barrier, so effective in
APML with CNS involvement.
-Less myelosuppresion.
-No need of further Anthracycline exposure.
44.
Newer targeted therapy
•Gemtuzumab ozogamicin- used in the western
countries, in combination with ATRA plus ATO in high
risk APML patients as well as relapsed patient proves
safe and effective.
• ATRA-ATO with minimal chemotherapy shows better
success rate to standard ATRA plus anthracycline .
• Tamibarotene (AM-80)- Tami-ATO combination
shows similar efficacy & less toxic than ATRA- ATO
• combo now used in Japan. .
45.
Case study
• Patient:R.M., 35 years, Female
• Ward/Clinic: Olive
• Date of Review: 24/10/2025
• Chief Complaints:
• - Easy fatigability
• - Menorrhagia (3 cycles)
• - Oral sores
• Past Medical History:
• - Known hypertension (since 2018) after childbirth
• - No current documentation of antihypertensive therapy