W-Curve & Perl
•
1 like•13,729 views

How I adapted the W-Curve analysis tool for DNA for automated use using Perl.

Recommended
More Related Content
Similar to W-Curve & Perl
Similar to W-Curve & Perl (20)
More from Workhorse Computing
More from Workhorse Computing (20)
Recently uploaded
Recently uploaded (20)
W-Curve & Perl
- 1. Throwing a Wcurve: WholeGenome Analysis in Perl ● Steven Lembark <slembark@cheetahmail.com> http://www.bioinformatics.org
- 2. A short description of the Wcurve wholegenome comparison project ● A really quick description of why genome comparison is useful and messy – and why the Wcurve is interesting. ● How I adapted a graphical display algorithm to make use of Perl and BioPerl. ● A few tricks for bulk data analysis in Perl: triangular comparison using stable metrics and hash slices from integer sequences.
- 3. One of the biggest advances in science was sequencing genes. ● Genes provide the blueprint for life, and are the core of new medicine and technology. ● Drugs are being developed to cure diseases where only symptoms could be treated before. ● Bioinformatics is core of a new kind of biology that can process genetic information in ways unimagined only 10 years ago.
- 4. We did not evolve to be computable. ● Comparing genes is difficult. ● Genes are written in called our DNA as sequences of “bases” labeled “C”, “A”, “T” and “G”. ● The genes mostly generate proteins, which are made of twenty amino acids. ● The genetic code is redundant and varies even within an individual; there is “junk” between the genes and within them; along with variable “repeat” groups.
- 5. Redundant Coding ● The triplets are called Leu Arg L R UUA, CGU, UUG, CGC, CUU, CGA, CUC, CUA, CUG CGG, AGA, AGG “Codons”, and actually Ser S UCU, UCC, UCA, UCG, AGU, AGC Val V GUU, GUC, GUA, GUG Pro P CCU, CCC, CCA, CCG encode RNA (with Ala A GCU, GCC, GCA, GCG Thr T ACU, ACC, ACA, ACG bases of C, A, G, & U). Gly G GGU, GGC, GGA, GGG Ile I AUU, AUC, AUA Lys K AAA, AAG ● The 64 combinations of Asn N AAU, AAC Asp D GAU, GAC RNA encode only 20 Phe F UUU, UUC Cys C UGU, UGC protein building blocks. Gln Glu Q E CAA, GAA, CAG GAG His H CAU, CAC ● This makes “equality” a Tyr Y UAU, UAC Met M AUG slippery question Trp W UGG between genes. Start AUG, CUG, UUG, GUG, AUU Stop UAG, UGA, UAA
- 6. What a difference a base makes... ● The difference between Normal and Sickle Cell Hemoglobin is caused by a point mutation: one differing DNA base changing an amino acid. Normal gtt cat tta Sickle Cell gtt gtt tta gtc cac tta gtc gtg tta ● Replace any sequence on the left gta cat tta gtg gtt ctc gtc cac ttg gtt gta cta with any on the right and you gtg cat ctc gta gta tta gtt cac cta gtg gtg ctg have Sickle Cell Anemia. gtg cac ctg gtc cac ttg gta cac ctt gta gtt ctt ● This difference is among 450_000 ... bases.
- 7. Exonic DNA and repeats ● Much of our DNA produces RNA that is edited out before protein transcription. ● Exons are the DNA sequence that actually encodes a protein. ● Even “standard” exonic genes have bits of extra material in them called repeats: O, A, B blood types happen because varying number of repeated “TA” sequences cause slightly different proteins to result. ● This means that two “normal” copies of hemoglobin may also differ only by having multiple copies of some filler DNA.
- 8. WholeGenome Comparisons ● Evolutionary biology and drug research both try to compare all of one organism to another in search of commonality for evolutionary history or odds that a disease or cure may be common to the species. ● This adds to our problems the variability between species along with all of the withinspecies (or individual) variation I've shown so far. ● People have two hemoglobin genes, which can vary between them: genome comparisons also most accommodate variances within individuals.
- 9. Not quite a consensus ● For comparing textbook genetics, the “Consensus” sequence helps remove some variability. ● This only helps when comparing reviewed sequences that have one: newly discovered sequences or the raw output of sequencing equipment will be in whatever order the organism really has – with all of its variability intact. ● In fact, one use of these comparison techniques is determining if different encodings are simply variations on the consensus.
- 10. Comparing Genes ● Our bodies gracefully deal with variability in genes thousands of times a second; unfortunately for Bioinformatics, computers deal with this much more slowly. ● The common approaches to comparing genes are Alignment, Hidden Markov Models, and Graphical. ● Alignment uses recursive algorithms to find what does match; HMM's look at probabilities that they match; graphical models map the problem onto something that supports approximation.
- 11. Traditional gene matching: Alignment ● Traditional method is alignment: BLAST & FASTA are the standards here. ● They line up the portions of the sequence, leaving gaps as necessary. ● Recursion necessary to shift the mapped portions makes these slow and them to a few thousand bases. ● Alignment studies require significant manual intervention to set up the comparison process.
- 12. Waiting in line for a gene: Hidden Markov Models ● Hidden Markov Models (“HMM”) generate a state transition model from one set of DNA used to train a model, then estimate the probability that another sequence is from the same family. ● These are slow to train and exquisitely sensitive to the choice of DNA sequence used for training. ● They may require more DNA sequences for training than are readily available, leading to smallsample error or skewed results.
- 13. Graphical Models ● Graphical models abstract the genetic code into some n dimensional space for comparison. Geometric algorithms can then be used to analyze or compare the curves. ● These are largely intended to use the human brain to perform the comparison. ● 3D models add dimensions that allow for approximate results and greater freedom in the algorithms used to compare genes. ● The Wcurve uses a 3D model, with a simple state machine generating the curves.
- 14. The WCurve Code ● The original layout was designed by a Java programmer for use in displaying DNA for visual comparison. ● It was slow and nearly useless for computed comparison. ● My job was to fix it using – of course – Perl. ● The rest of this talk describes what I went through, both in Perl and the algorithm itself, to get a workable comparison technique.
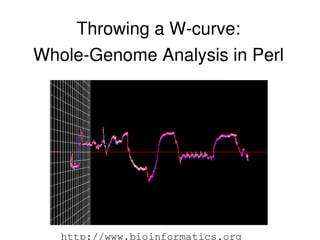
- 15. The WCurve Algorithem ● The basic design is a state machine crawling down the DNA sequence. ● Each corner of a square is associated with one type of DNA base. ● The curve is generated by moving from the current location half way to the corner associated with the next base.
- 16. Improving the WCurve ● First thing I had to do was find a measure amenable to comparing the curves; then improve the algorithm for computing them. ● Our goal was to find a fast process for wholegenome comparison. ● This meant being able to load DNA, generate curves, and compare them quickly without manual intervention. ● The result described here is an fast, heuristic utility which can be developed to perform more exact comparisions with different measures.
- 17. Approximate Mesure ● The comparison rules must accommodate small differences between sequences. ● I used the difference along the longer vector's length: this ignores small differences and adds the two lengths when the vectors point in opposite directions (A > 90 degrees). ● The measure for comparing two genes is the average of their differences over the length of the longer gene with [0,0] filler on the shorter one.
- 18. Computing the Wcurve ● Now all I had to do was compute and compare the curves quickly enough. ● This involved changing the coordinate system to cylindrical, redesigning the statebox, hashing the computed curves by length, and finding efficient ways to compare the arrays. ● I also took into account some knowledge about the DNA, including the need to differentiate AT and CGrich regions of a sequence.
- 19. Cylindrical Coords ● The original cartesian coordinates made halfintervals easy to compute but complicated computing the difference measure. ● Changing the code to use cylindrical notation (r, angle, Z) simplified comparing the curves, but left the distances computed using the square root of two (distance of origin to (1,1)style corners). ● This would have caused significant accumulated error along the full length of a gene.
- 20. Initial fixes: Modify the Curve ● Rotating the square so that it's corners were on the axis simplified the computations and avoided the rounding error. ● Putting AT and CG on common edges leaves the curve less likely to hug the origin. ● The angle to a corner (“A”) is simply a matter of adding multiples of PI/2 from a table. ● The half interval to a corner is simply: ( 1 + r1 * cos(A) ) / ( 2 * cos(A/2) ) with a simple check for 2 * cos(A/2) == 0
- 21. Next: Computing Curves in Perl ● Single curves can easily be stored as arrays, the catch is finding efficient ways to generate them. ● Given an array of DNA and another of Wcurve, one of them can be handled via forloop iterator, but the other requires an index or a shift to walk down. ● C handles these situations via pointers; Perl requires a bit more finesse.
- 22. Compute wcurves in place ● The good news was that once a Wcurve point was computed its DNA base was used up and could be discarded. ● This left me able modify $_ with the result of computing on $_ to construct the curves in place. This code replaces each letter of the DNA sequence with its curve point: my @curve = split //, $dna; my $state = [ 0, 0 ]; $_ = generate_w_curve $state, $_ for @curve; $seqz{ $name } = @curve;
- 23. Comparing Lengths: Arrays ● Another issue was comparing genes in groups by length. Genes with base counts (or DNA string lengths) more than 10% different will rarely be the same gene. ● The simple approach is to store them by length in an array: push @{$curvz[$len]}, $curve; ● Access to the lengths would be an array slice of @curvz[ 0.90*$len .. 1.10*$len ]; ● Problem here is dealing with a long (Hemoglobin is 450_000 bases) sparse array.
- 24. Comparing Lengths: Hashes ● Large, sparse lists are better handled by hashes. ● This left me with @curvz{ (0.90*$len .. 1.10*$len ) } ● Using a numeric range operator to generate hash keys works just fine: Perl will happily convert your numeric lists into strings for hash access. ● That leaves me with nested hashes of ref's to scalars. The outer key is a length, the inner key a gene name, the leaf value a wcurve.
- 25. Uppertriangular comparisons ● If A == B imiplys B == A, only half of the comparisons need to be made. ● The issue for Wcurves was making sure that the same comparison was done regardless of the curve order. ● Instead of comparing the length of the first curve I ended up using the longer one to compute the measure, with [0,0] filler in the shorter curve. ● This left me with @curvz{ $len .. 1.1 * $len }
- 26. Now all I needed was DNA... ● Genbankformat files have full genomes but are complicated to parse – their format is regexproof. ● Bioperl (and Lincoln Stein ) solved that one for me, using IO objects. ● The main problem with Bioperl is – due to parallel development with other Bio* packages – it looks way too much like Java in many cases; down to the point of requiring 34 opaque objects to do anything, each of which has its own fairly opaque documentation. ● In the end I was able to read each .gbk file and write its genes back out in FASTA format for comparison.
- 27. Extracting data from .gbk files sub read_genome Bio::SeqIO handles { ● # grab a copy of the local genbank file # as a Bio::SeqIO. the only useful thing the guts of a # # from it are the features whose primary tag is a gene. Genbank file use Bio::SeqIO; gracefully. my @seqargz = ( qw( -format genbank -file ), shift ); ● The result is a species my $fh = Bio::SeqIO->new( @seqargz); my $seq = ( $fh->next_seq )[0]; name followed by my ( $species ) an arrayref feature = $seq->{species}->common_name =~ m{^(S+s+S+)}; objects. ( $species, [ grep { $_->primary_tag eq 'gene' } $seq->get_SeqFeatures ] ) }
- 28. Extracting the ID and Sequence sub gene_sequences What I need from { ● # first step: slurp the genes only. the objects are my ( $species, $genome ) = read_genome shift; the gene name # now map the names onto their sequences. # caller gets back anonymous hash of the and exonic # gene names mapped onto their sequences. (“spliced”) DNA. my $gene_seqz = { map { ● Once they were ( $_->get_tag_values('gene'), extracted the ) $_->spliced_seq->seq BioSeq object } @$genome }; could be # at this point the genome and SeqIO objects discarded. # can be discarded: all we need going # forward is the the text handed back here. ( $species, $gene_seqz ) }
- 29. Output as FASTA for my $path ( @ARGV ) ● The the outer loop { # snag the species name and dna string. simply cycles the my ( $species, $genome ) = gene_sequences $path; Genbank files, ( my $base = $species ) =~ s/s+/_/g; writing out each while( my($gene,$seq) = each %$genome ) gene as a FASTA { my $path = “$Bin/../var/$base.$gene.fasta"; file. open my $fh, '>', $path; ● Aside: this can # matching on 1,80 char's breaks the long # string up into separate lines; newlines # via $, easily be forked print $fh by input file. “> $input, $species, $gene", '', $seq =~ /.{1,80}/g; } }
- 30. Example FASTA output ● The resulting FASTA file has minimal information on the '>' line, with the file sorted by size for more efficient processing: > U00089.gbk, Mycoplasma pneumoniae, yfiB ATGCAAGATAAAAACGTCAAAATTCAGGGCAATCTGGTACGGGTACACCTTTCGGGATCGTTTCTGAAGTTCCAGGCAAT TTACAAGGTGAAAAAGCTGTACTTACAGCTGTTAATTCTCTCCGTGATTGCCTTCTTTTGGGGCTTGTTAGGAGTTGTGT TTGTCCAGTTTTCTGGATTATATGACATTGGCATTGCTTCCATTAGTCAGGGCTTAGCACGGTTAGCGGATTATTTAATT AGGTCGAACAAGGTCAGTGTGGATGCTGACACCATTTACAACGTCATCTTCTGGTTGAGTCAAATTCTGATTAACATTCC CTTATTTGTTTTGGGTTGGTACAAGATTTCCAAAAAGTTTACCTTGTTAACCCTTTACTTTGTGGTAGTCTCCAACGTTT TTGGGTTTGCCTTCTCTTACATTCCGGGCGTGGAAAACTTCTTCTTGTTTGCTAATTTAACTGAACTTACTAAGGCCAAC GGTGGCTTAGAACAAGCGATTAACAACCAAGGGGTGCAACTGATCTTTTGGGAACAAACCGCTGAAAAGCAAATTTCGTT AATGTTCTATGCGCTGATCTGGGGTTTTCTTCAAGCTGTGTTTTACTCAGTTATCCTAATTATTGATGCATCGAGTGGTG GGTTGGACTTTTTGGCCTTCTGGTATTCGGAAAAGAAACACAAGGACATTGGTGGTATTTTGTTTATTGTTAACACCCTT AGTTTCTTGATCGGTTACACCATTGGCACTTACCTTACCGGTAGCTTACTAGCACAAGGCTTTCAAGAAGATAGACAAAA ACCGTTTGGAGTGGCTTTTTTCTTGTCCCCTAACTTAGTGTTTACGATTTTCATGAACATTATCTTAGGGATCTTTACCT CCTACTTCTTTCCTAAATACCAGTTTGTCAAAGTGGAAGTGTATGGTAAACACATGGAACAAATGCGCAACTACTTGTTG AGCAGTAACCAGTCCTTTGCGGTCACTATGTTCGAAGTGGAAGGGGGGTACTCGCGCCAAAAGAACCAGGTGTTAGTTAC AAACTGTTTGTTTACGAAAACGGCCGAACTTTTAGAAGCTGTTAGACGAGTCGATCCGGATGCTCTGTTCTCAATTACCT TCATTAAAAAGTTGGATGGTTATATCTATGAAAGAAAAGCACCTGATAAAGTAGTCCCACCA GTAAAAGACCCAGTTAAAGCCCAGGAAAATTAA
- 31. Storing DNA for comparison ● Catch: the whole genome of anything more than bacteria won't fit into memory at one time. ● Since I didn't need all of the DNA in memory at once, so I could store a hash of { length }{ geneid } that was false until it was first processed, setting ref $_ || $_ = generate_curve $_ as each item was being processed. ● I was also able to delete usedup lengths as they were processed.
- 32. Performing the comparisons ● Back to the issue of iterating two arrays again. ● Linked lists are not used often in Perl but this is one case they really apply: advancing the two nodes requires only: ( $node, $r, $a ) = @$node ● The only other issue was avoiding rounding errors computing 2*cos($a/2). ● At the edge of precision the value can be nonzero but still yield essentially infinite results. ● The fix was to set the value using: $value = 0 if $value < $TINY;
- 33. Result: Wcurve output For comparison: This took 45 hours of computing time to validate with FASTA at NIH. Whole Gnome Comparison:Mycoplasma genitalium, Mycoplasma pneumoniae Curve Description: Curve Used: WCurve with T A G C Score Cutoff: 0.3 Length Cutoff: 0.15% Report Size: Base Genes: 480 Matched Base genes: 72 15% Report Rows: 72 15% Filter Efficiency: Cartesian Product: 330240 Alt. Genes Compared: 28851 8.73% Total Comparisons: 44020 13.32% Time Efficiency: Elapsed time: 565 sec Comparison Time: 558 sec 98% Comparison Rate : 78 Hz Results By Gene Row Mycoplasma genitalium Mycoplasma pneumoniae Score 1 MG325 rpmG 0.158080075467006 2 MG362 rplL 0.176481395732838 3 MG451 tuf 0.185903240607304 4 MG197 rpmI 0.204703167254187 ...
- 34. Summary: Perly Data Handling ● You may not need all of the data in memory all of the time. ● Breaking I/O up into chunks often helps: multiple pagesize reads are more efficient than a single large slurp. ● Preprocess data saves sorting, chunking during processing. ● Symmetric tests cut the number of comparisons by half. ● Use $_ to replace data in place rather than store both inputs and outputs. ● Look at your computations: simply rotating a box can help.