Download as PDF, PPTX
![m (2.4) that the VCP depends not only on the actualy oxygen concentration
riences, but also on the total number of cells. In the following sections we
ach of these factors individually, beginning with the number of cells within
which we will call the carrying capacity.
ygen dependence
d the changes in the radiation parameters, α and β, with oxygen we use the
oxygen enhancement ratio (OER):
αi =
αmax
OERαi
(pi)
, βi =
βmax
OERβi
(pi)2
, (2.5)
nd βmax are the values of α and β under fully oxygenated conditions and
re the values of α, β and oxygen, p, in compartment i. We can further find
function of the oxygen concentration by using the relation established by
correlation by Chapman et al. [22], and Palcic and Skarsgard [62]:
OERi =
(OERmax − OERmin)Km
pi + Km
+ OERmin (2.6)
oxygen concentration in compartment i, Km = 3.28 and OERαmin =OERβmin =
There is little information about how β changes in glioblastoma with oxy-
o for now, we will assume that the α
β
ratio for maximally sensitive cells
ant at 10 Gy−1
(this is reported in the literature ranging from 8.64 [25] to
31
Oxygen dependence
and the changes in the radiation parameters, α and β, with oxygen we use the
the oxygen enhancement ratio (OER):
αi =
αmax
OERαi
(pi)
, βi =
βmax
OERβi
(pi)2
, (2.5)
x and βmax are the values of α and β under fully oxygenated conditions and
pi are the values of α, β and oxygen, p, in compartment i. We can further find
s a function of the oxygen concentration by using the relation established by
tal correlation by Chapman et al. [22], and Palcic and Skarsgard [62]:
OERi =
(OERmax − OERmin)Km
pi + Km
+ OERmin (2.6)
the oxygen concentration in compartment i, Km = 3.28 and OERαmin =OERβmin =
6]. There is little information about how β changes in glioblastoma with oxy-
n, so for now, we will assume that the α
β
ratio for maximally sensitive cells
nstant at 10 Gy−1
(this is reported in the literature ranging from 8.64 [25] to
31
he basics of radiobiological modeling, to include tumor control
ear-quadratic model of surviving fraction (SF) [15, 31], the
ement ratio (OER) and linear energy transfer (LET).
SF = e−n(αd+βd2)
(2.1)
nd β refer to the radiobiologic parameters associated (phe-
kill secondary to ‘single hit’ events (α) and ‘double hit’ events
r fraction of radiation and n to the number of fractions.
trol probability
e that considers the total number of surviving cells in a tumour,
apter, in the total number of surviving cells in our domain. We
sure, based on the TCP, which we will call the Voxel Control
derstand the effect of radiation on a distribution of cells, we
vival probability of each discrete subpopulation of cells in the
ed by their proliferative state, and their microenvironmental
allow Nt
ij to be the number of cells of type i, where i ∈ {S, D}
30
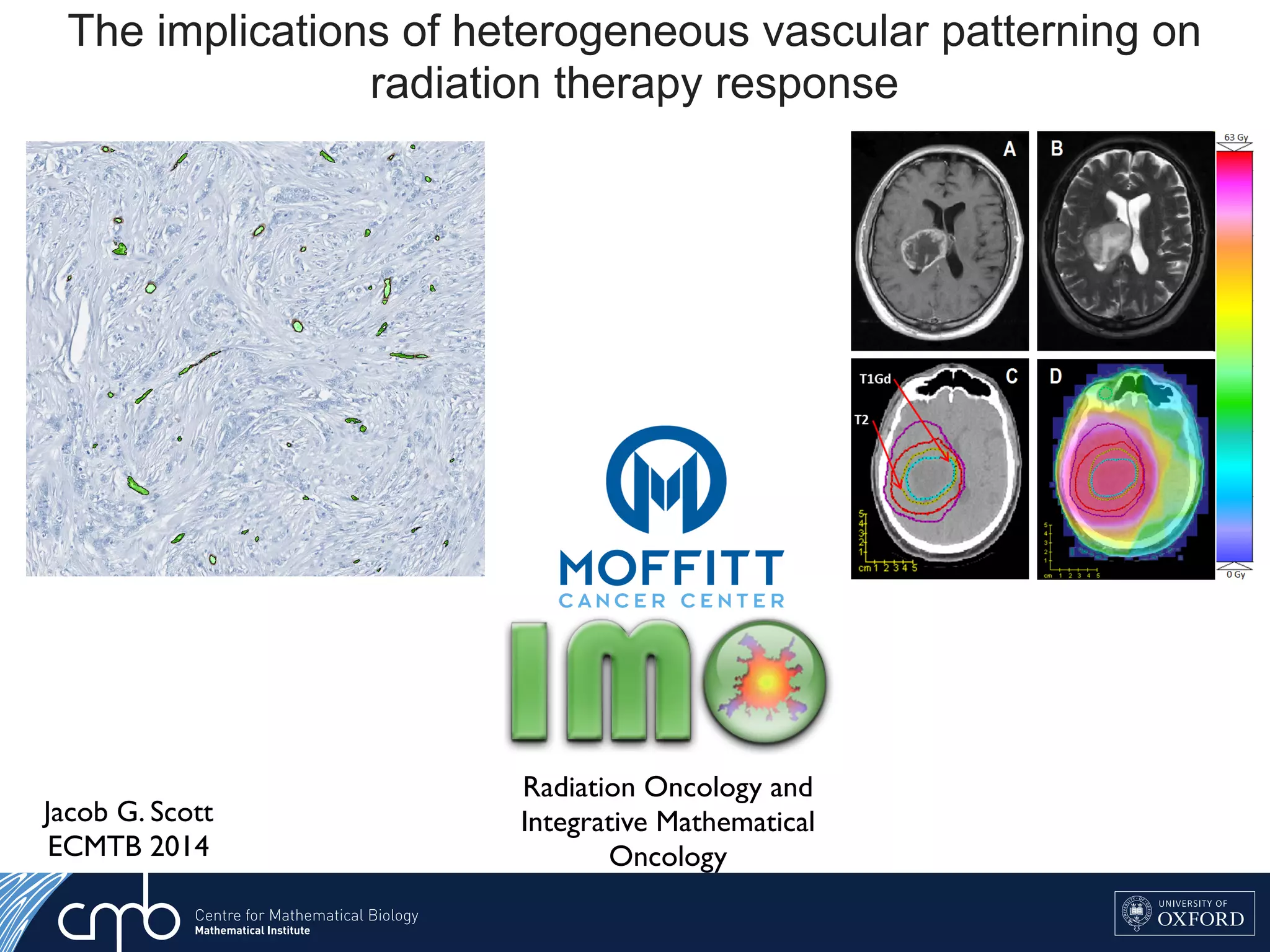
image credit: http://www.eyephysics.com/tdf/models.htm
Radiation Biology Primer
TCP = e−SFN0](https://image.slidesharecdn.com/ecmtb2014vascularpatterning-140623120030-phpapp02/75/Ecmtb2014-vascular-patterning-4-2048.jpg)
![∂c(x, t)
∂t
= Dc∇2
c(x, t) − fc(x, t), (1.1)
entration of oxygen at a given time t and position x, Dc is the
ygen, which we assume to be constant (providing linear, isotropic
governed by Michaelis-Menten kinetics and is defined as:
µir(c, t) if there is a cell of type i at x at time t,
0 otherwise,
(1.2)
nd the labels H, S, P and T are used to refer to healthy, TIC, TAC
y. Here µi is defined as the cell type-specific oxygen consumption
T ), which modulates r(c, t), the oxygen dependent consumption
r(c, t) = rc
c(x, t)
c(x, t) + Km
e the maximal uptate rate and effective Michaelis-Menten con-
upplement equation (1.1) with the following initial and boundary
ith the oxygen in the domain set to c(x, 0) = c0 and all lattice
mal cells. In the case of a cancer simulation, we replace the center
e TIC. Vessels are placed throughout the domain at a prescribed
5
nt of oxygen equal to that carried in the arterial blood. This oxygen is then allowed
ffuse into the surrounding tissue.
he spatiotemporal evolution of the oxygen field is described by the reaction-diffusion
al differential equation (PDE)
∂c(x, t)
∂t
= Dc∇2
c(x, t) − fc(x, t), (1.1)
e c(x, t) is the concentration of oxygen at a given time t and position x, Dc is the
ion coefficient of oxygen, which we assume to be constant (providing linear, isotropic
ion) and fc(x, t) is governed by Michaelis-Menten kinetics and is defined as:
fc(x, t) =
µir(c, t) if there is a cell of type i at x at time t,
0 otherwise,
(1.2)
e i ∈ {H, S, P, T} and the labels H, S, P and T are used to refer to healthy, TIC, TAC
TD cells respectively. Here µi is defined as the cell type-specific oxygen consumption
ant (µH, µS, µP , µT ), which modulates r(c, t), the oxygen dependent consumption
defined as
r(c, t) = rc
c(x, t)
c(x, t) + Km
e rc and Km denote the maximal uptate rate and effective Michaelis-Menten con-
respectively. We supplement equation (1.1) with the following initial and boundary
tions. We begin with the oxygen in the domain set to c(x, 0) = c0 and all lattice
s occupied by normal cells. In the case of a cancer simulation, we replace the center
e point with a single TIC. Vessels are placed throughout the domain at a prescribed
5
source of oxygen occupying one lattice point, are placed randomly
ce at the start of a given simulation, with a specified spatial density
we neglect vascular remodelling. Each vessel is assumed to carry an
ual to that carried in the arterial blood. This oxygen is then allowed
rrounding tissue.
ral evolution of the oxygen field is described by the reaction-diffusion
quation (PDE)
∂c(x, t)
∂t
= Dc∇2
c(x, t) − fc(x, t), (1.1)
concentration of oxygen at a given time t and position x, Dc is the
f oxygen, which we assume to be constant (providing linear, isotropic
) is governed by Michaelis-Menten kinetics and is defined as:
=
µir(c, t) if there is a cell of type i at x at time t,
0 otherwise,
(1.2)
} and the labels H, S, P and T are used to refer to healthy, TIC, TAC
vely. Here µi is defined as the cell type-specific oxygen consumption
P , µT ), which modulates r(c, t), the oxygen dependent consumption
r(c, t) = rc
c(x, t)
c(x, t) + Km
note the maximal uptate rate and effective Michaelis-Menten con-
We supplement equation (1.1) with the following initial and boundary
n with the oxygen in the domain set to c(x, 0) = c0 and all lattice
ormal cells. In the case of a cancer simulation, we replace the center
ingle TIC. Vessels are placed throughout the domain at a prescribed
ll diameters [33] and the information from the literature concerning the ratio of
r to normal oxygen consumption (see Section 1.2.2.1).
ntroducing the non-dimensional variables ˜x = x/L, ˜t = t/τ and ˜c = c/c0, we define
ew non-dimensional parameters
˜Dc =
Dcτ
L2
, ˜rc =
τn0rc
c0
. (1.3)
otational convenience, we henceforth drop the tildes and refer to the non-dimensional
meters only as DC and rc. See Table 1.1 for a full list of parameter estimates and
ndix ?? for our procedure for esimating the cancer cell oxygen consumption rate.
.3 Numerical solution
der to solve equation (1.1) numerically, we discretize space and time by considering
k∆t, xi = i∆x and yj = j∆x and approximate the concentration of oxygen at
tep k and position (i∆x, j∆x) by ck
i,j ≈ c(xi, yj, tk). We use a central difference
oximation for the Laplacian and thus approximate equation (1.1) by
ck+1
i,j − ck
i,j
∆t
=
DC
∆x2
ck
i+1,j + ck
i−1,j + ck
i,j+1 + ck
i,j−1 − 4ck
i,j
−
fc
k
i,j
, (1.4)
e
fc
k
i,j
is the cell-specific oxygen consumption µcellrc at time k given a cell at
on (i∆x, j∆x) as discussed in equation 1.2. We then rearrange equation (1.4) to
n a solution for ck+1
i,j , yielding
rc
Maximal oxygen
consumption rate
2.3 × 10−16
mol cells−1
s −1
[32]
c0
Background oxygen
concentration
1.7 × 10−8
mol cm −2
[6]
∆x
Average cell
diameter
50µm [25]
τ
Average cell
doubling time
16h [19]
cap
Hypoxic
threshold
0.1 [20]
rp
Proliferative oxygen
consumption
5 × rc [32]
Km
Effective Michaelis-
Menten constant
0.8mmHg [54]
n0
Cancer cell
density
1.6 × 105
cells cm −2
[21]
s
TIC symmetric
division probability
0 ≤ s ≤ 1 Model-specific
a
TAC proliferative
capacity
0 − 10 Model-specific
µcancer/µH
Cancer metabolic
ratio
2 [12]
ck+1
i,j = ck
i,j
1 − 4
DC∆t
∆x2
+
DC∆t
∆x2
ck
i+1,j + ck
i−1,j + ck
i,j+1 + ck
i,j−1
− ∆t(fc)k
i,j. (1.5)
During each update, then, the oxygen tension in a given lattice point is updated
Diffusion and uptake, exact
Diffusion and uptake, numeric approximation](https://image.slidesharecdn.com/ecmtb2014vascularpatterning-140623120030-phpapp02/75/Ecmtb2014-vascular-patterning-5-2048.jpg)
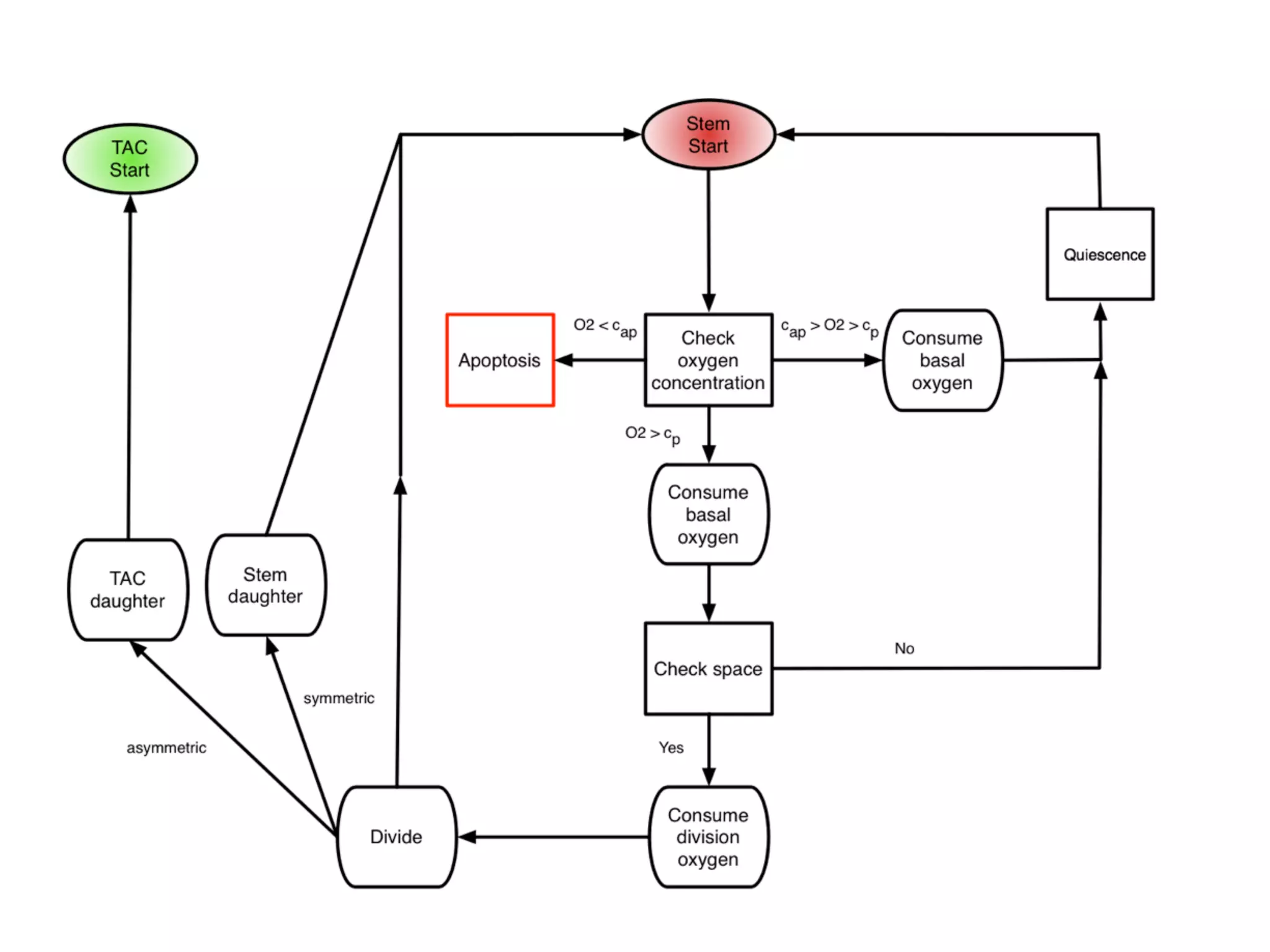
![autophagy (directly translated as ‘self-eating’), a state in which they become resistant
to nutrient starvation [91], and cells are known to die on different time scales and by
different mechanisms (apoptosis vs. necrosis) depending on the magnitude and duration
of the hypoxic insult. While these differences have been shown to affect tumour growth
[19], as this is not the main aim of this model, we will simplify this scenario by assigning
a rate, pd, for cell death at each cellular automaton update defined in Section 2.2.3, when
under extreme hypoxia (i.e. c cap).
2.2.2.4 Quiescence
When cells sense that there is not enough oxygen to divide, or experience contact inhi-
bition, they undergo a state of quiescence during which there is no division. We model
this as an oxygen threshold (c cp) below which cellular division is not possible and by
the spatial constraint which requires the cell to be quiescent if there is not at least one
neighbouring lattice point (Moore neighbourhood, see Figure 2.7(a)) empty or inhabited
by a normal cell. When a cell senses that sufficient oxygen and a neighbouring site have
become available, the state of quiescence is reversed.
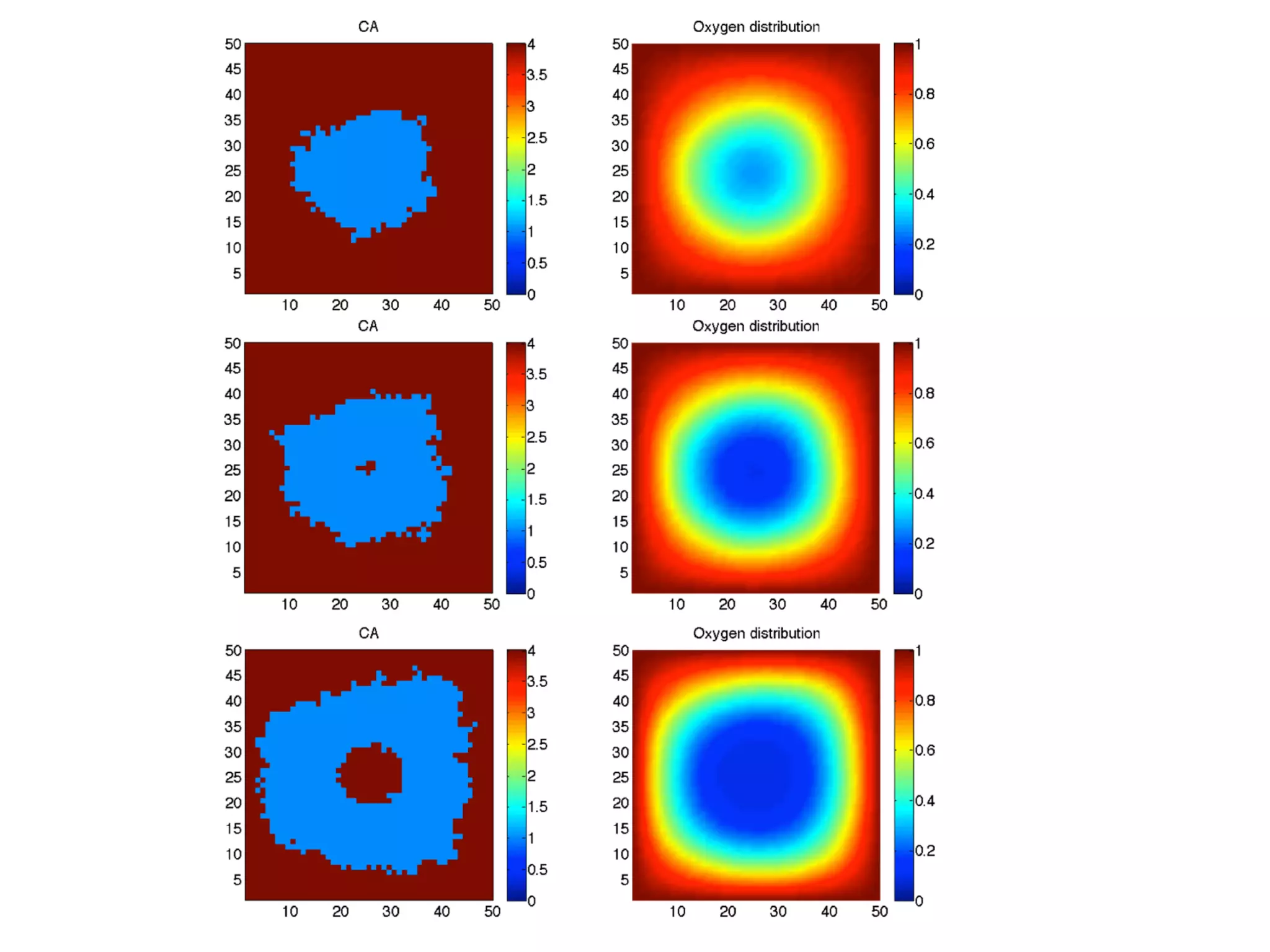
Figure 2.3: A summary of oxygen based cell fate threshholds. At each cellular au-
tomaton update, each cell in the domain undergoes a series of fate decisions based on the local
oxygen concentration. When c c , cells die at rate p , when c c c cells are quiescent](https://image.slidesharecdn.com/ecmtb2014vascularpatterning-140623120030-phpapp02/75/Ecmtb2014-vascular-patterning-6-2048.jpg)


![∂c
∂t
= D
∂2
c
∂x2
(1.6)
[0, L], with boundary conditions c(0, t) = c(0, L) = 1 and and initial
0.
this equation using separation of variables and solving for ˜c = c + 1
) and the boundary conditions ˜c(0, t) = ˜c(0, L) = 0 and initial condition
find that the solution (see Appendix A) is given by
) =
∞
n=1
2
nπ
cos nπ − 1
sin
nπx
L
exp
−
n2
π2
DCt
L2
. (1.7)
we plot the oxygen concentation profile through the centre of a 100×100
evoid of cells at one-second intervals. The analytical solution (equation
d to the numerical solution of equation (1.5) at each lattice point for
, 500 and 1, 000 seconds. We find excellent agreement between the two
cales and updates
time scales that govern the diffusion of nutrients and that at which
anaged by updating the continuous part of the model many times per
This can become computationally expensive in this explicit scheme,
seek to minimize this number. However, for stability, we require that
∂c
∂t
= D
∂2
c
∂x2
(1.6)
for t ≥ 0 and x ∈ [0, L], with boundary conditions c(0, t) = c(0, L) = 1 and and initial
condition c(x, t) = 0.
We can solve this equation using separation of variables and solving for ˜c = c + 1
which satisfies (1.6) and the boundary conditions ˜c(0, t) = ˜c(0, L) = 0 and initial condition
˜c(x, t) = −1. We find that the solution (see Appendix A) is given by
c(x, t) =
∞
n=1
2
nπ
cos nπ − 1
sin
nπx
L
exp
−
n2
π2
DCt
L2
. (1.7)
In Figure 1.8 we plot the oxygen concentation profile through the centre of a 100×100
domain which is devoid of cells at one-second intervals. The analytical solution (equation
(1.7)) is compared to the numerical solution of equation (1.5) at each lattice point for
time t = 1, 10, 100, 500 and 1, 000 seconds. We find excellent agreement between the two
solutions.
1.2.3.4 Time scales and updates
The difference in time scales that govern the diffusion of nutrients and that at which
cells operate is managed by updating the continuous part of the model many times per
cellular time step. This can become computationally expensive in this explicit scheme,
and therefore, we seek to minimize this number. However, for stability, we require that
the ∆tDc/∆x2
term from equation (1.5) is less than 0.25 [56]. We therefore choose
∆tDc/∆x2
= 0.1, which equates to updating oxygen every 0.25 seconds, or approximately
230, 400 times per cell cycle based on the parameters chosen (see Table 1.1). While we
assume the average cell cycle time to be τ = 16 hours, it is well known that cells in tissues
are not synchronized, and also that cell fate decisions such as apoptosis are made on
shorter time scales. To model this heterogeneity in division time and to more accurately
match the finer time scale associated with cell death due to microenvironmental cues
∂c
∂t
= D
∂2
c
∂x2
(1.6)
for t ≥ 0 and x ∈ [0, L], with boundary conditions c(0, t) = c(0, L) = 1 and and initial
condition c(x, t) = 0.
We can solve this equation using separation of variables and solving for ˜c = c + 1
which satisfies (1.6) and the boundary conditions ˜c(0, t) = ˜c(0, L) = 0 and initial condition
˜c(x, t) = −1. We find that the solution (see Appendix A) is given by
c(x, t) =
∞
n=1
2
nπ
cos nπ − 1
sin
nπx
L
exp
−
n2
π2
DCt
L2
. (1.7)
In Figure 1.8 we plot the oxygen concentation profile through the centre of a 100×100
domain which is devoid of cells at one-second intervals. The analytical solution (equation
(1.7)) is compared to the numerical solution of equation (1.5) at each lattice point for
time t = 1, 10, 100, 500 and 1, 000 seconds. We find excellent agreement between the two
solutions.
1.2.3.4 Time scales and updates
The difference in time scales that govern the diffusion of nutrients and that at which
cells operate is managed by updating the continuous part of the model many times per
cellular time step. This can become computationally expensive in this explicit scheme,
and therefore, we seek to minimize this number. However, for stability, we require that
the ∆tDc/∆x2
term from equation (1.5) is less than 0.25 [56]. We therefore choose
∆tDc/∆x2
= 0.1, which equates to updating oxygen every 0.25 seconds, or approximately
230, 400 times per cell cycle based on the parameters chosen (see Table 1.1). While we
assume the average cell cycle time to be τ = 16 hours, it is well known that cells in tissues
are not synchronized, and also that cell fate decisions such as apoptosis are made on
shorter time scales. To model this heterogeneity in division time and to more accurately
match the finer time scale associated with cell death due to microenvironmental cues
∂c
∂t
=
for t ≥ 0 and x ∈ [0, L], with boundary con
condition c(x, t) = 0.
We can solve this equation using separa
which satisfies (1.6) and the boundary condit
˜c(x, t) = −1. We find that the solution (see
c(x, t) =
∞
n=1
2
nπ
cos nπ − 1
In Figure 1.8 we plot the oxygen concenta
domain which is devoid of cells at one-second
(1.7)) is compared to the numerical solutio
time t = 1, 10, 100, 500 and 1, 000 seconds. W
solutions.
1.2.3.4 Time scales and updates
The difference in time scales that govern t
cells operate is managed by updating the co
cellular time step. This can become compu
and therefore, we seek to minimize this num
the ∆tDc/∆x2
term from equation (1.5) i
∆tDc/∆x2
= 0.1, which equates to updating
230, 400 times per cell cycle based on the p
assume the average cell cycle time to be τ =
are not synchronized, and also that cell fa
shorter time scales. To model this heterogen
match the finer time scale associated with
[68], we choose to update the HCA 100 t
∂c
∂t
= D
∂ c
∂x2
(1.6)
for t ≥ 0 and x ∈ [0, L], with boundary conditions c(0, t) = c(0, L) = 1 and and initial
condition c(x, t) = 0.
We can solve this equation using separation of variables and solving for ˜c = c + 1
which satisfies (1.6) and the boundary conditions ˜c(0, t) = ˜c(0, L) = 0 and initial condition
˜c(x, t) = −1. We find that the solution (see Appendix A) is given by
c(x, t) =
∞
n=1
2
nπ
cos nπ − 1
sin
nπx
L
exp
−
n2
π2
DCt
L2
. (1.7)
In Figure 1.8 we plot the oxygen concentation profile through the centre of a 100×100
domain which is devoid of cells at one-second intervals. The analytical solution (equation
(1.7)) is compared to the numerical solution of equation (1.5) at each lattice point for
time t = 1, 10, 100, 500 and 1, 000 seconds. We find excellent agreement between the two
solutions.
1.2.3.4 Time scales and updates
The difference in time scales that govern the diffusion of nutrients and that at which
cells operate is managed by updating the continuous part of the model many times per
cellular time step. This can become computationally expensive in this explicit scheme,
and therefore, we seek to minimize this number. However, for stability, we require that
the ∆tDc/∆x2
term from equation (1.5) is less than 0.25 [56]. We therefore choose
∆tDc/∆x2
= 0.1, which equates to updating oxygen every 0.25 seconds, or approximately
230, 400 times per cell cycle based on the parameters chosen (see Table 1.1). While we
assume the average cell cycle time to be τ = 16 hours, it is well known that cells in tissues
are not synchronized, and also that cell fate decisions such as apoptosis are made on
shorter time scales. To model this heterogeneity in division time and to more accurately
match the finer time scale associated with cell death due to microenvironmental cues
Check of numerics](https://image.slidesharecdn.com/ecmtb2014vascularpatterning-140623120030-phpapp02/75/Ecmtb2014-vascular-patterning-23-2048.jpg)

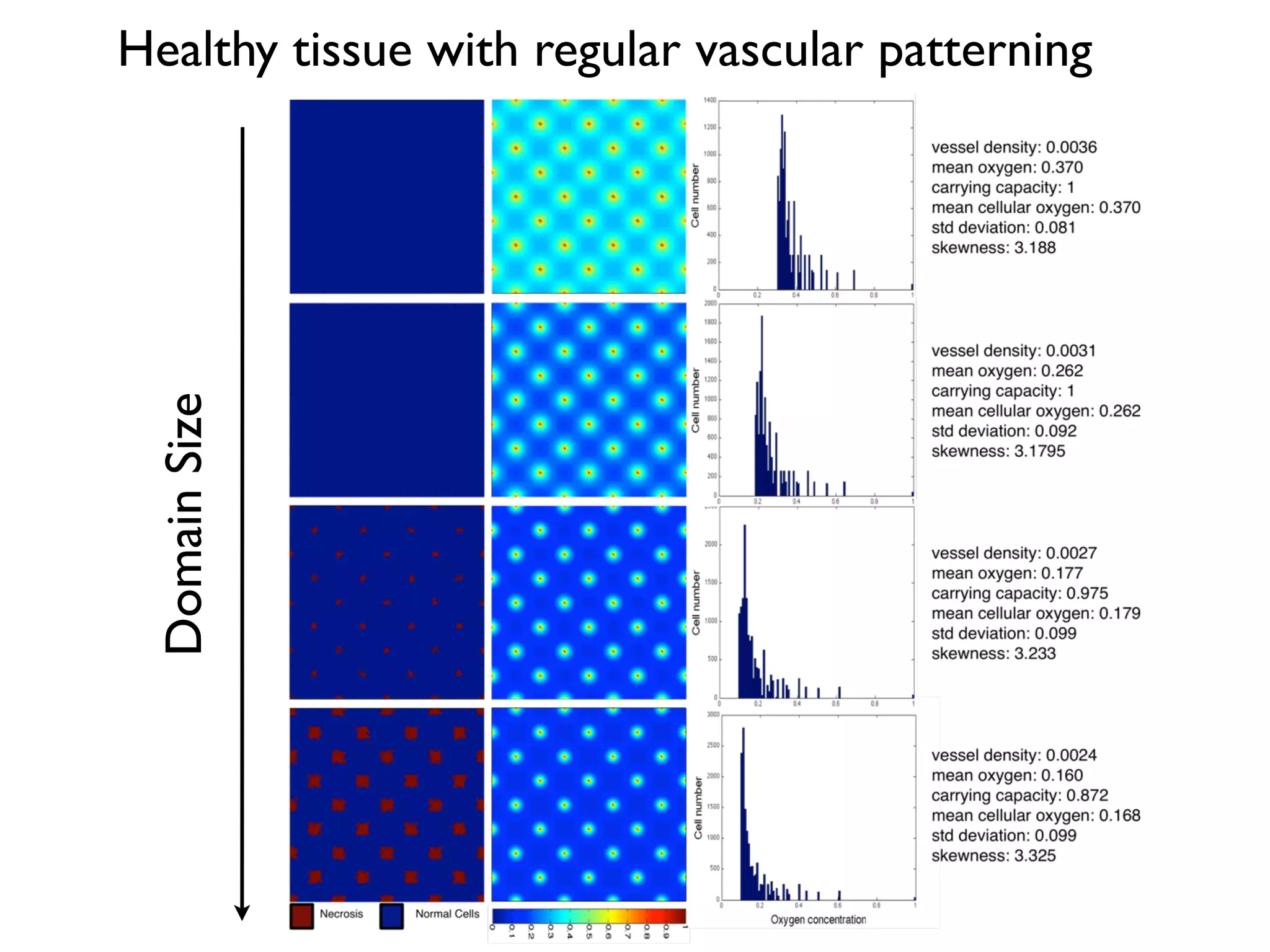
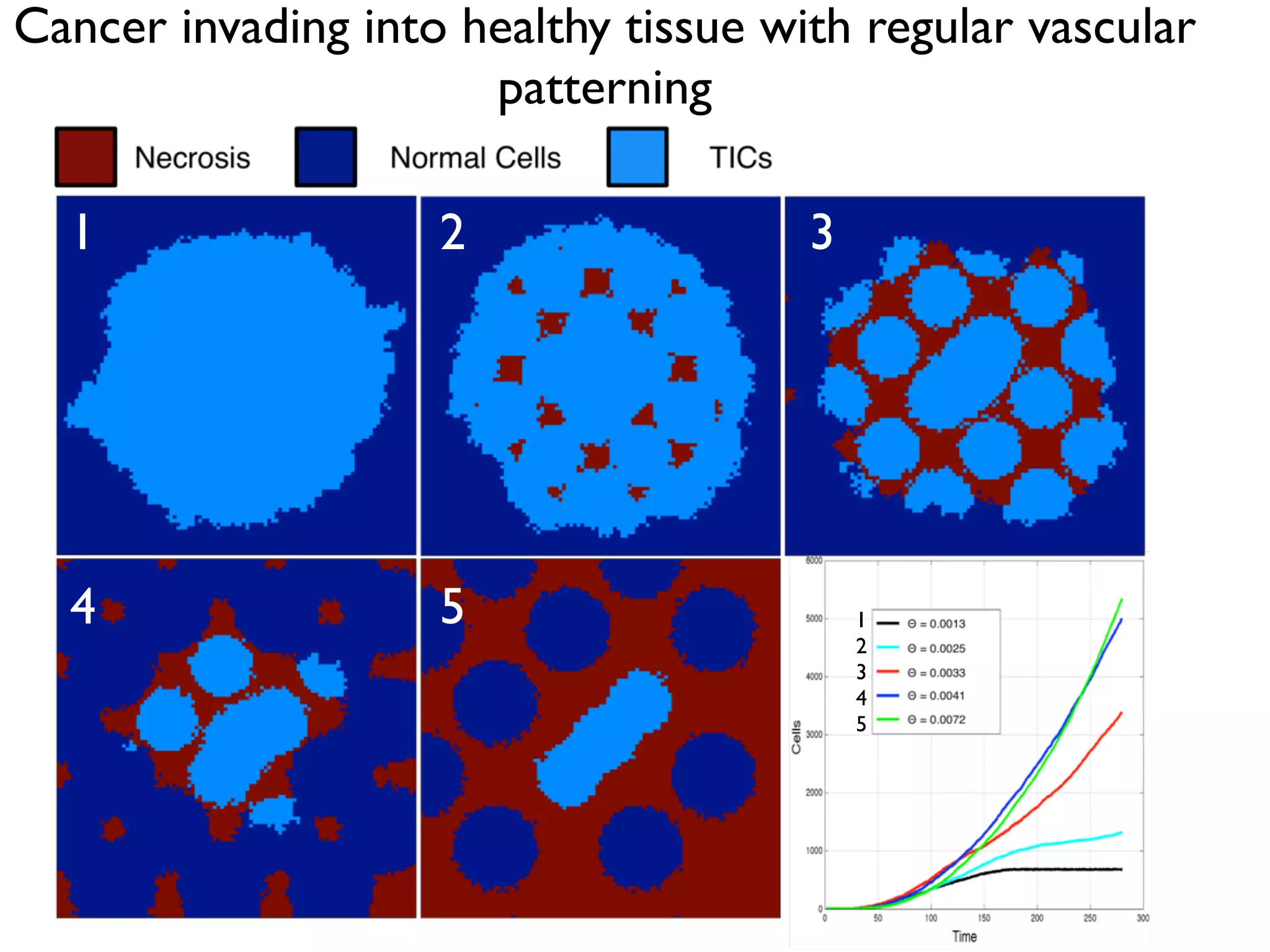
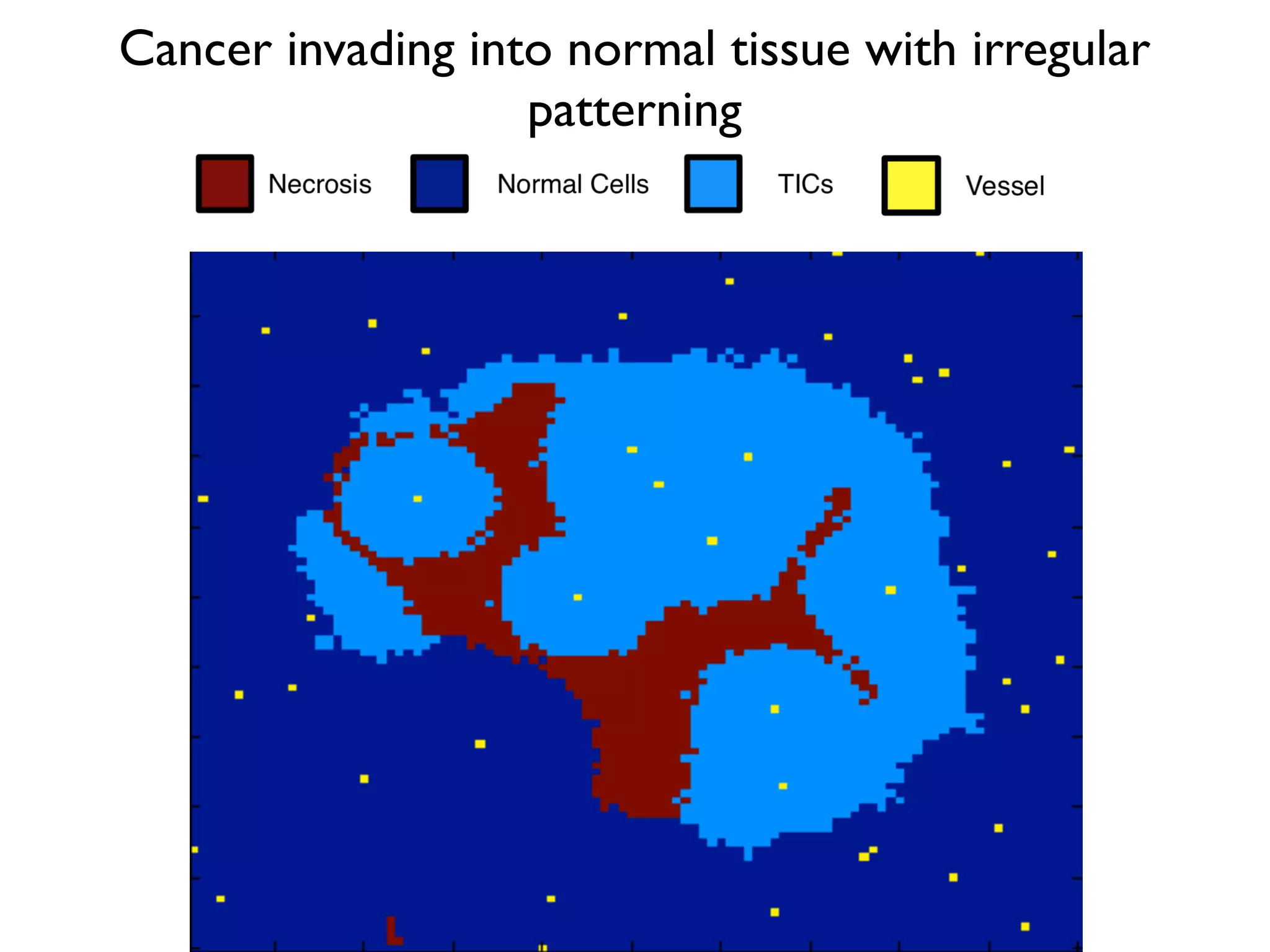
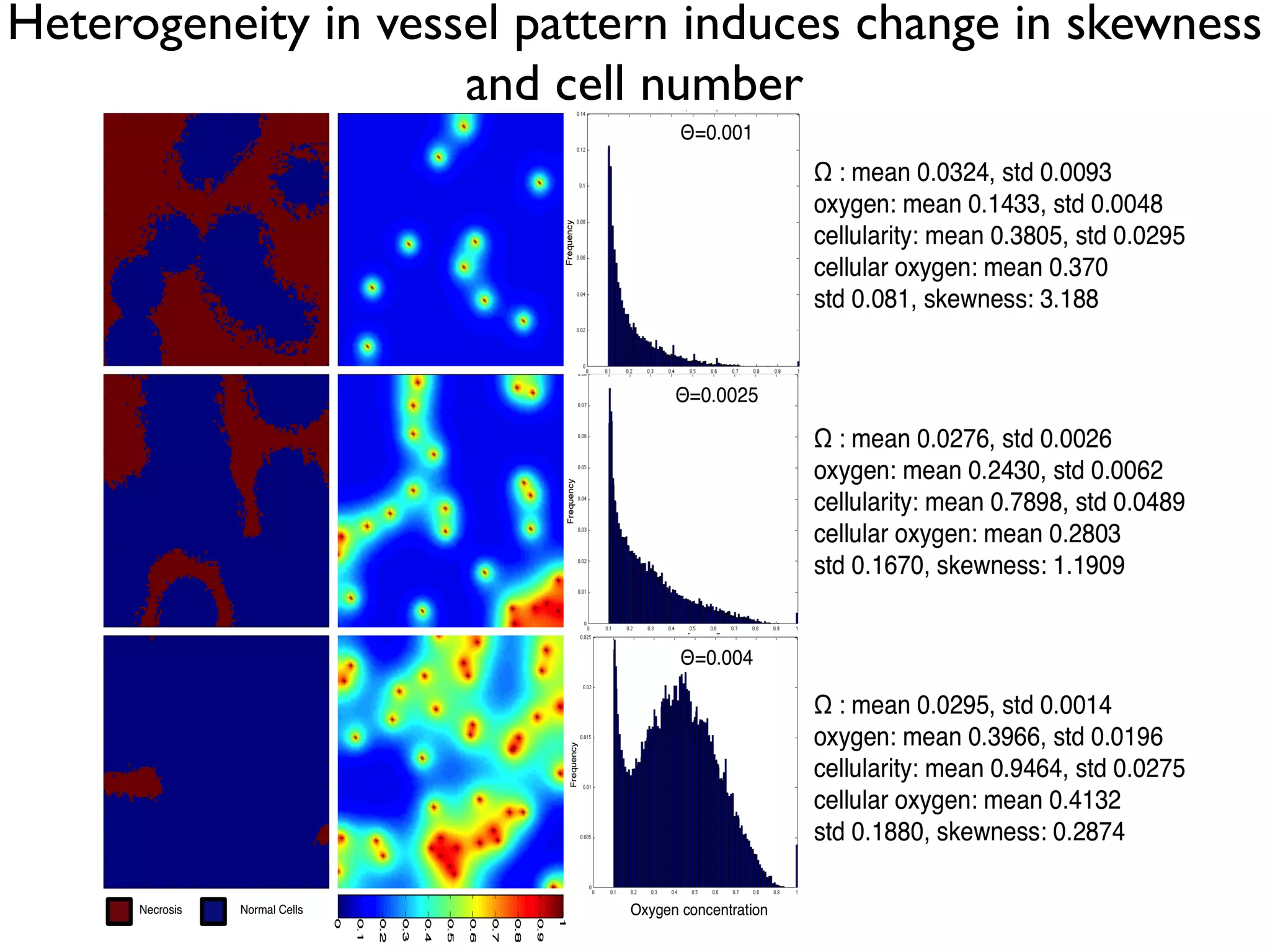
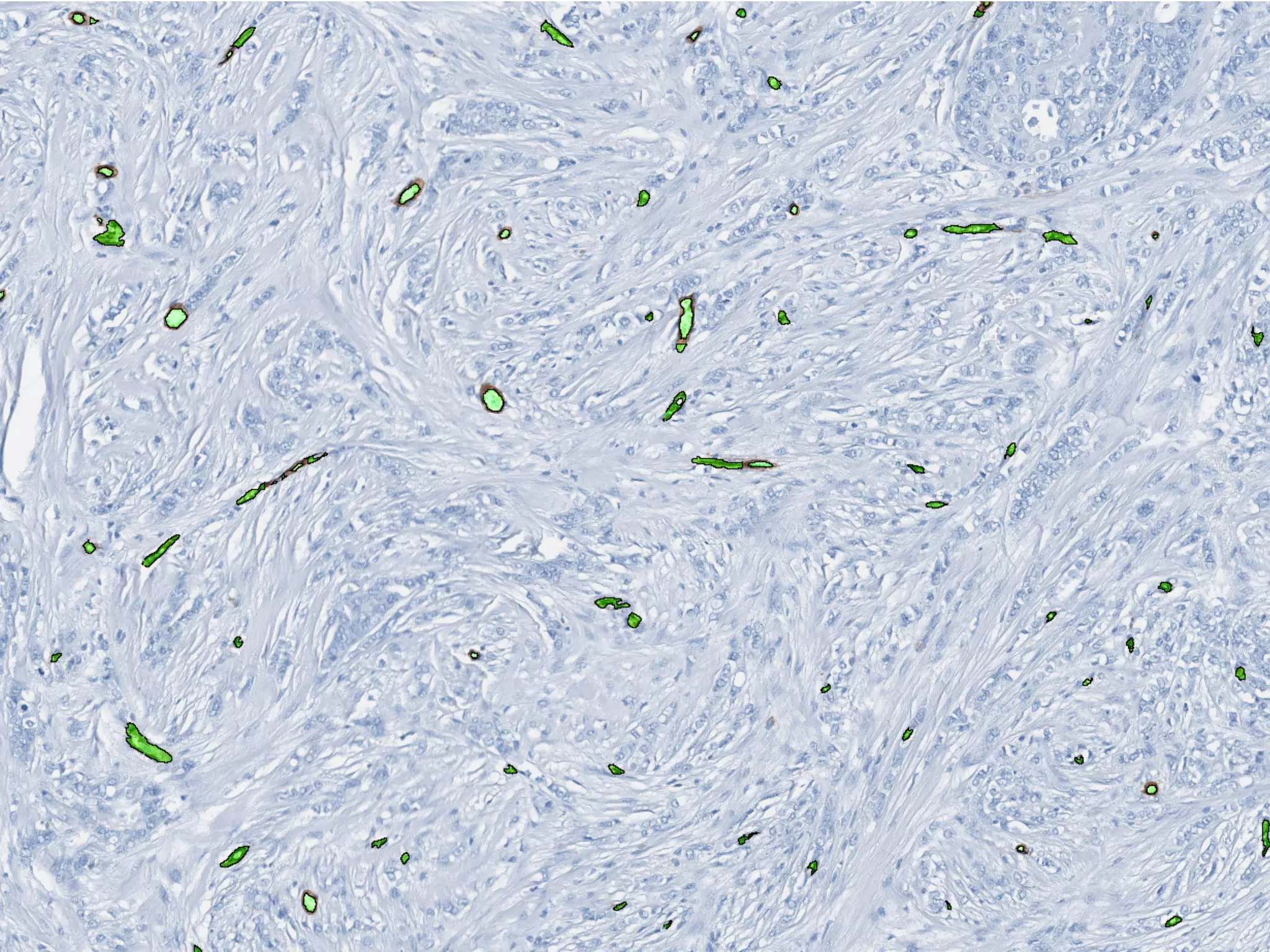
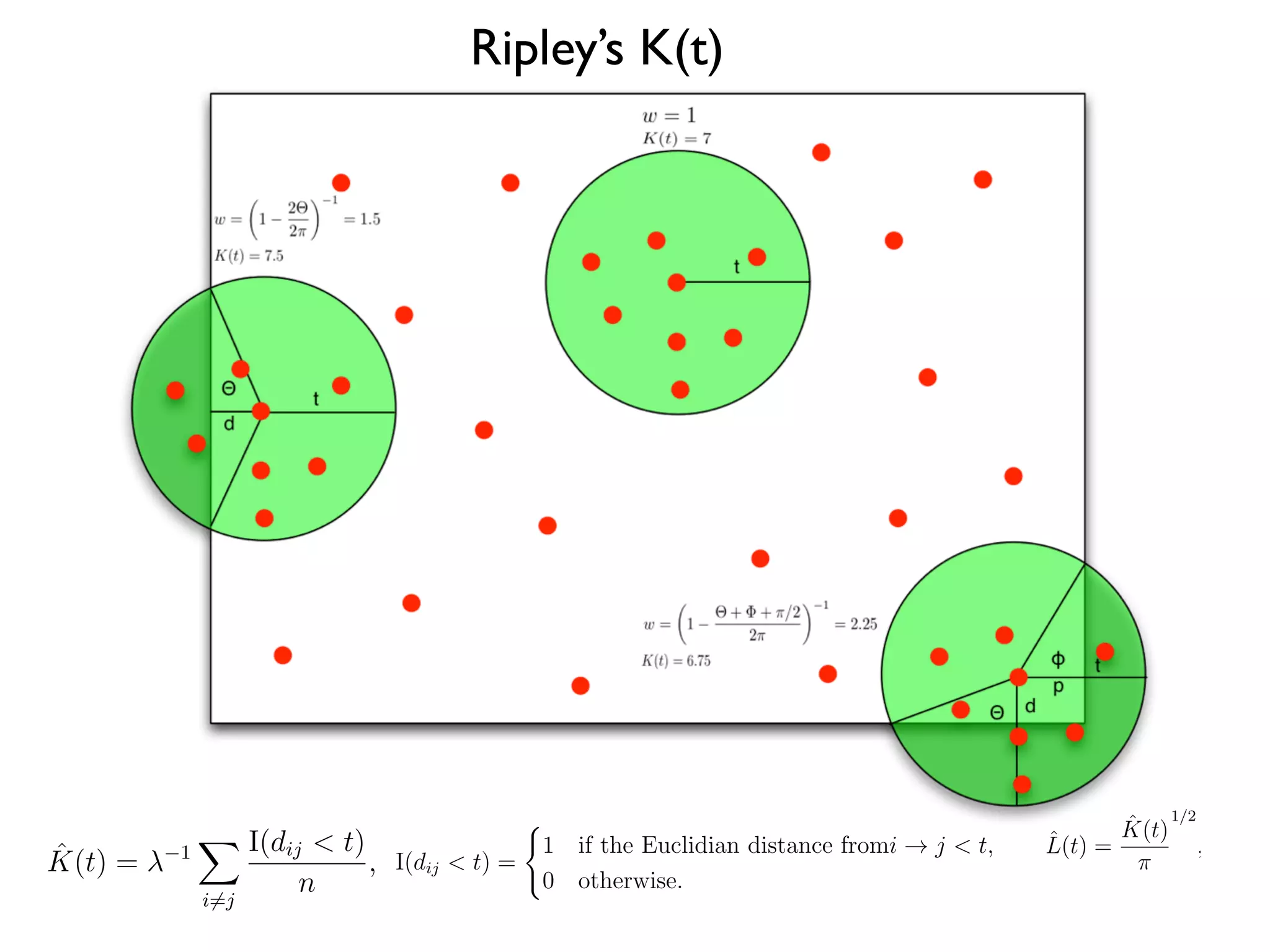
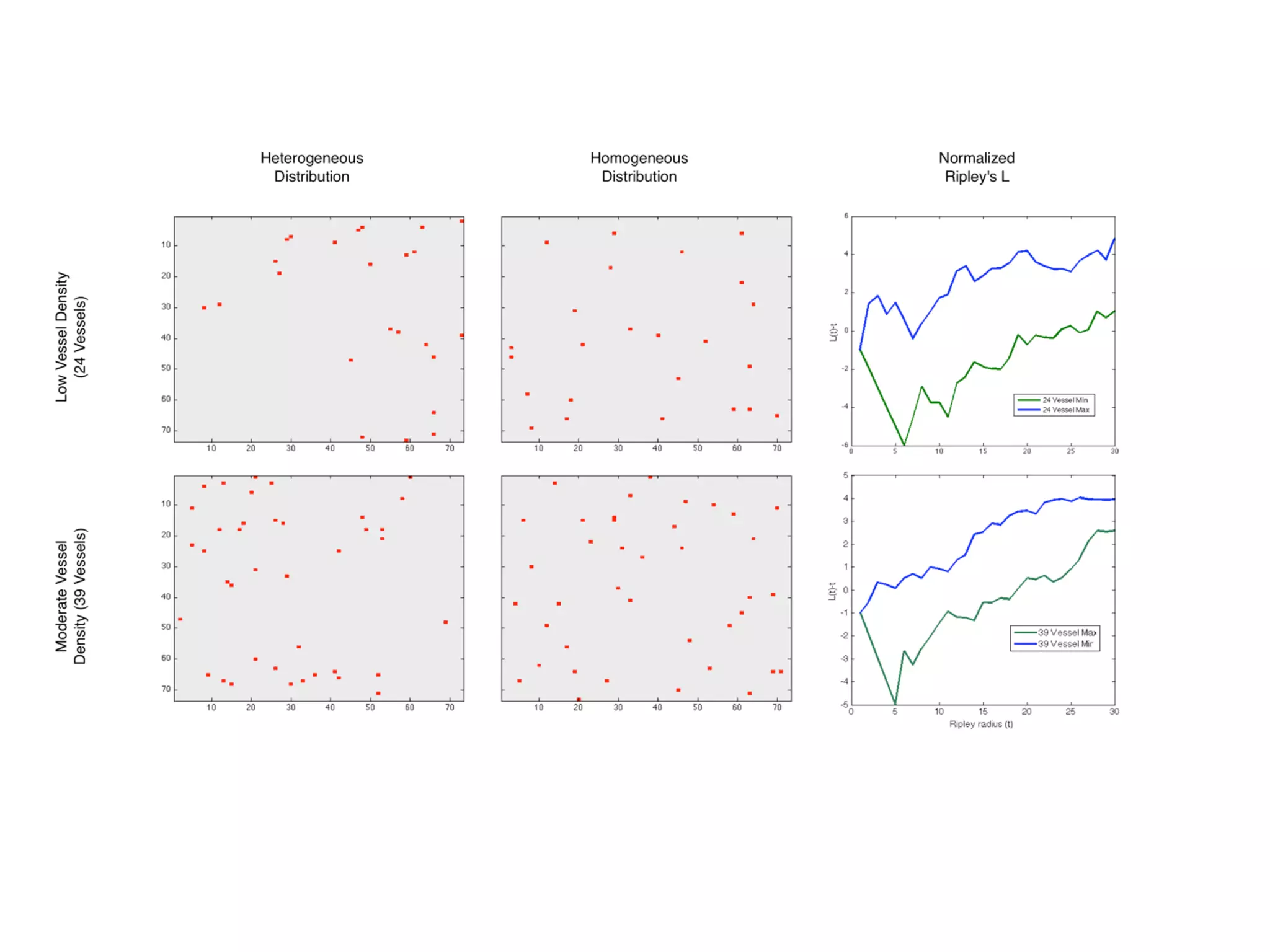
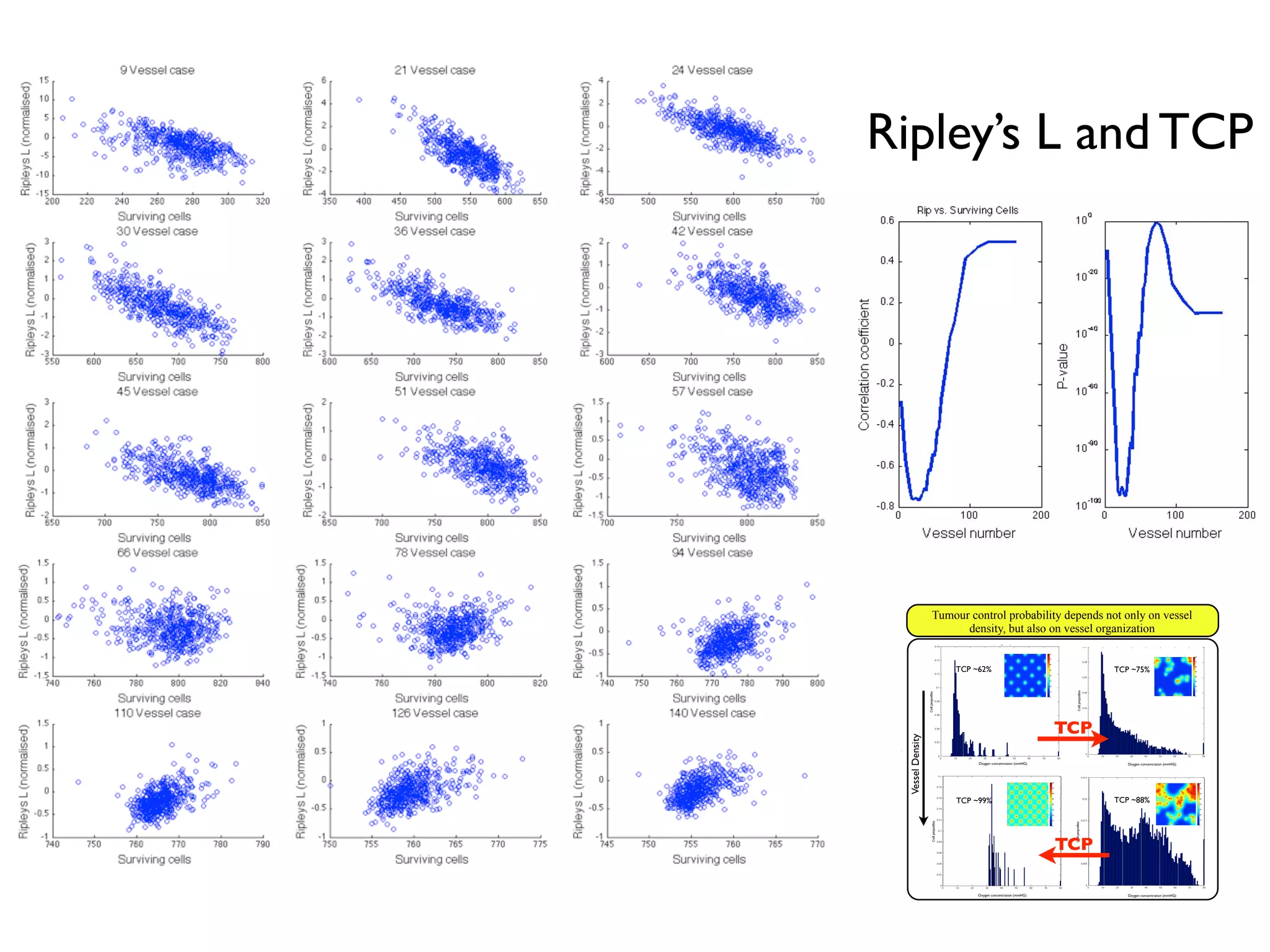
1. The document discusses a computational model that simulates tumor growth and response to radiation therapy. It accounts for heterogeneous vascular patterning in tissue which can impact oxygen distribution, cell behavior, and treatment response. 2. The model represents tissue at a microscopic scale using a lattice-based cellular automaton. It models oxygen diffusion and consumption, as well as cell proliferation, migration, quiescence and death based on local oxygen levels. 3. The model can incorporate patient-specific vessel patterns reconstructed from medical images to simulate their effects on tumor oxygenation and radiation therapy outcomes at a personalized level. This could help optimize treatment plans accounting for tissue-level heterogeneity.















































