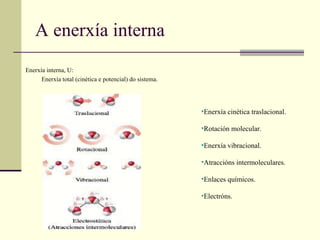
A enerxía internaEnerxía interna, U: Enerxía total (cinética e potencial) do sistema. Enerxía cinética traslacional. Rotación molecular. Enerxía vibracional. Atraccións intermoleculares. Enlaces químicos. Electróns.
5.
A Calor Acalor é a enerxía que se intercambia entre un sistema e os seus arrededores como resultado dunha diferenza de temperaturas. A calor flúe desde o corpo máis quente ata o corpo máis frío
6.
Unidades para ocalor e o traballo Caloría (cal): É a cantidade de calor necesaria para variar un grao Celsius a temperatura dun gramo de auga. Xulio (J): É a unidade de enerxía básica para o calor no Sistema internacional (S.I) 1 cal = 4,184 J
As variables termodinámicasNa termodinámica, a descrición do estado dun sistema realízase mediante os valores de determinadas propiedades macroscópicas denominadas variables termodinámicas , tales como P, V, T, m, ... Non tódalas variables son independentes, chega coñecer os valores dun pequeno número delas para caracterizar o sistema. Estas variables independentes denomínanse variables de estado . Toda función que poda expresarse con axuda das variables de estado denomínase función de estado do sistema. Exemplo: Enerxía interna (U), entalpía (H), Temperatura (T), Volume (V), Presión (P) etc
9.
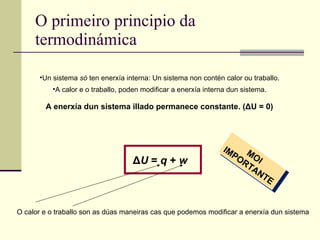
O primeiro principioda termodinámica Un sistema só ten enerxía interna: Un sistema non contén calor ou traballo. A calor e o traballo, poden modificar a enerxía interna dun sistema. Δ U = q + w A enerxía dun sistema illado permanece constante. ( Δ U = 0) MOI IMPORTANTE O calor e o traballo son as dúas maneiras cas que podemos modificar a enerxía dun sistema
O primeiro principioda termodinámica Δ U = q + w Vamos a fixarnos en cada un dos termos traballo (w) e calor (q)
12.
O traballo: compresión– expansión (traballo presión – volume) w = F x d = ( P x A ) x h = P V w = - P V Existe varios tipos de traballo: Eléctrico, mecánico ou de compresión - expansión. Nos tan só nos fixaremos no traballo de expansión e compresión
13.
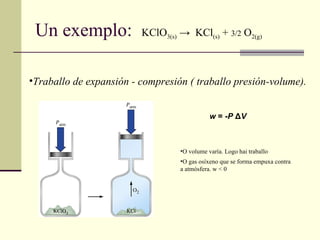
Un exemplo: Traballo de expansión - compresión ( traballo presión-volume). O volume varía. Logo hai traballo O gas osíxeno que se forma empuxa contra a atmósfera. w < 0 KClO 3(s) -> KCl (s) + 3/2 O 2(g) w = - P Δ V
Calor a volumeconstante (Q v ) É o intercambio de enerxía nun recipiente pechado que non cambia de volume. Se V = constante, é dicir, Δ V = 0 entón W = 0 e como Δ U = q + w Entón Qv = Δ U A calor á volume constante e igual a variación de enerxía interna MOI IMPORTANTE
16.
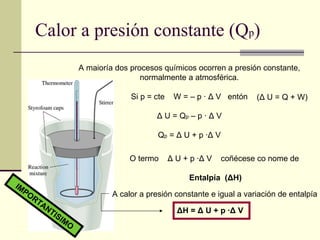
Calor a presiónconstante (Q p ) O termo Δ U + p · Δ V coñécese co nome de Entalpía ( Δ H) Δ H = Δ U + p · Δ V A calor a presión constante e igual a variación de entalpía IMPORTANTISIMO A maioría dos procesos químicos ocorren a presión constante, normalmente a atmosférica. Si p = cte W = – p · Δ V entón Δ U = Q p – p · Δ V Q p = Δ U + p · Δ V ( Δ U = Q + W)
17.
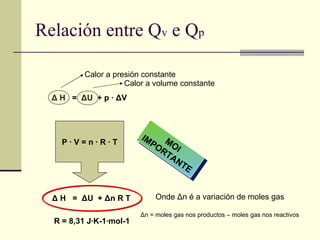
Relación entre Qv e Q p MOI IMPORTANTE Δ H = Δ U + p · Δ V Δ H = Δ U + Δ n R T P · V = n · R · T Calor a presión constante Calor a volume constante Onde Δ n é a variación de moles gas Δ n = moles gas nos productos – moles gas nos reactivos R = 8,31 J·K-1·mol-1
18.
Diferenza entre Δ H e Δ U q V = - 563,5 kJ/mol = U q P = - 566 kJ/mol = H Se a reacción anterior a volume constante é exotérmica, ¿ a calor desprendida a presión constante será maior ou menor ? CO (g) + ½ O 2 (g) CO 2 (g) Un exemplo: Fai os exercicios 1, 2 e 3 da páxina 3 e o 4 e 5 da páxina seguinte
19.
Entalpía: diagramas entálpicos H = H (productos) – H (reactivos) H = calor emitido ou absorbido durante unha reacción a presión constante H productos < H reactivos H < 0 H productos > H reactivos H > 0 É imposible coñecer a entalpía dunha substancia, pero non será difícil coñecer a súa variación.
20.
Entalpía das reaccións∆ Hº r Pero sería imposible tabular tódalas entalpías de tódalas reaccións. 2 H 2 (g) + O 2 (g) 2 H 2 O (l) CO (g) + 2 O 2 (g) CO 2 (g) + 2 H 2 O (l) CO 2 (g) + 2 H 2(g) CH 3 OH (l) ∆ Hº1= -571,6 kJ ∆ Hº2= -283 kJ ∆ Hº3= +726,5 kJ Para a realización dos exercicios de termoquímica, sería ideal poder dispoñer de tódalas entalpías de reacción, por exemplo: ..............................
21.
Entalpía estandar deformación, Definimos a entalpìa estándar de formación, ∆Hºf , “ como a variación de entalpìa ( Δ H) que ten lugar cando se forma un mol de composto a partir dos elementos que o constitúen, no seu estado de agregación máis estable en condicións estándar” ( 1 atm e 298 K ) . Para poder coñecer as variacións de entalpía necesitamos establecer un punto de referencia. Por definición asignase o valor 0 a os elementos no seu estado máis normal nas condicións estándar. Observa a táboa da páxina 5 Para solucionar o problema anterior: ∆ H º f H 0 (O 2 ) = 0 f H 0 (O 3 ) = 142 kJ/mol f H 0 (C, grafito) = 0 f H 0 (C, diamante) = 1.90 kJ/mol f
22.
Lei de Hess“ Cando uns reactivos se converten en produtos a variación de entalpía é a mesma independentemente de se a reacción ocorre nun só paso ou nunha serie de pasos consecutivos” Da cordo ca lei de Hess, as ecuacións termoquímicas poden tratarse como ecuacións alxébricas, así: Se invertemos o sentido dunha ecuación termoquímica o signo da entalpía cambia. Se multiplicamos unha reacción por un factor n, entón tamén deberemos multiplicar a entalpía polo mesmo factor
23.
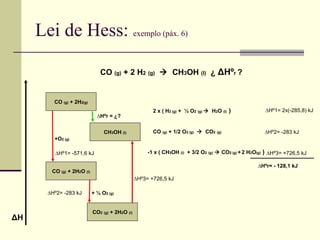
Lei de Hess: exemplo (páx. 6) CO (g) + 2 H 2 (g) CH 3 OH (l) ¿ Δ Hº r ? CO (g) + 2H 2(g) CH 3 OH (l) CO (g) + 2H 2 O (l) ∆ Hºr = ¿? CO 2 (g) + 2H 2 O (l) +O 2 (g) + ½ O 2 (g) ∆ Hº3= +726,5 kJ ∆ Hº2= -283 kJ ∆ Hº1= -571,6 kJ Δ H 2 x ( H 2 (g) + ½ O 2 (g) H 2 O (l) ) CO (g) + 1/2 O 2 (g) CO 2 (g) -1 x ( CH 3 OH (l) + 3/2 O 2 (g) CO 2 (g) + 2 H 2 O (g) ) ∆ Hº1= 2x(-285,8) kJ ∆ Hº2= -283 kJ ∆ Hº3= +726,5 kJ ∆ Hºr= - 128,1 kJ
24.
Lei de HessUnha formulación matemática da lei sería: Que usaremos cando dispoñamos das entalpías estandar de formación (táboa) Outra posibilidade sería aplicar a lei de Hess pero empregando as entalpías de formación
25.
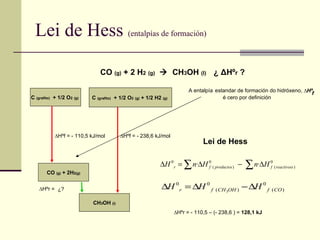
Lei de Hess (entalpías de formación) CO (g) + 2 H 2 (g) CH 3 OH (l) ¿ Δ Hº r ? CO (g) + 2H 2(g) CH 3 OH (l) ∆ Hºr = C (grafito) + 1/2 O 2 (g) C (grafito) + 1/2 O 2 (g) + 1/2 H2 (g) ∆ Hºf = - 110,5 kJ/mol ∆ Hºf = - 238,6 kJ/mol ¿? ∆ Hºr = - 110,5 – (- 238,6 ) = 128,1 kJ Lei de Hess A entalpía estandar de formación do hidróxeno, é cero por definición ∆ Hº f
26.
Entalpías de enlaceA partir das entalpías de enlace pódense estimar as entalpías de reacción Reacción química : : Proceso no que se rompen uns enlaces e fórmanse otros novos. Posto que a entalpía de enlace depende da molécula tabúlanse valores promedio ¡Ollo! * É un método aproximado ESTIMACIÓN Fiabilidade: 10 kJ * Só é aplicable a reaccións entre gases .
27.
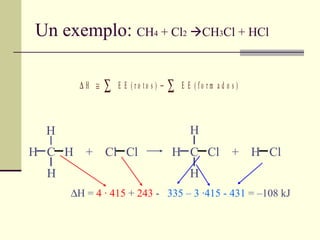
Un exemplo: CH 4 + Cl 2 CH 3 Cl + HCl H C H + Cl Cl H C Cl + H Cl H H H H H C H + Cl Cl H C Cl + H Cl H H H H H = 4 · 415 + 243 - 335 – 3 ·415 - 431 = –108 kJ
Temos tres maneirasde averiguar a entalpía dunha reacción Mediante as entalpías de formación. Combinando diferentes reaccións Mediante as entalpías de enlace. LEI DE HESS
30.
Calorimetría Q proceso = - Q desprendida ou absorbida Q absorbida ou desprendida = Q dos compoñentes + Q recipientes e aparellos Q proceso = - (Ce substancias · m substancias · T + Ce aparellos · m aparellos · Δ T ) Q proceso + Q desprendida ou absorbida = 0 A calor producida nun proceso transmítese tanto as substancias que interveñen como ao propio recipiente Q proceso = - Ce substancias · (m substancias + m eq )· T Q proceso = - Ce auga · (m substancias + m eq )· T Q = Ce · m · T
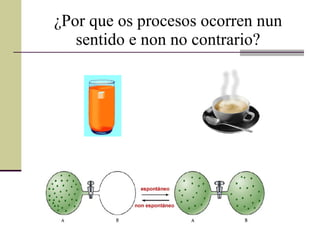
¿Por que osprocesos ocorren nun sentido e non no contrario?
33.
¿Por que ocorrenunhas reaccións e outras non? Diremos que unha reacción química é espontánea cando é un proceso permitido, e dicir, que unha vez comezado o proceso, pode continuar por si só. ¡¡¡ A espontaneidade non ten nada que ver ca velocidade !!!
34.
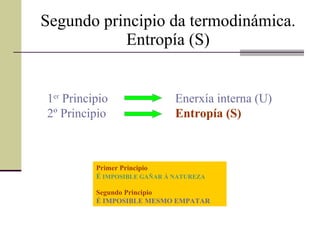
Segundo principio datermodinámica. Entropía (S) Nun principio creuse que a variación de entalpía ( Δ H) era un bo indicador da espontaneidade dunha reacción química, pois, tódalas reacción exotérmicas eran espontáneas Sen embargo coñécense procesos espontáneos e a vez endotérmicos, como por exemplo: a descomposición do carbonato de calcio ou mesmo o paso do xeo a auga entre outros. CaCO 3 CO 2 + CaO Δ H > 0 H 2 O sólido H 2 O líquido Δ H > 0
35.
Segundo principio datermodinámica. Entropía (S) Primer Principio É IMPOSIBLE GAÑAR Á NATUREZA Segundo Principio É IMPOSIBLE MESMO EMPATAR 1 er Principio Enerxía interna (U) 2º Principio Entropía (S)
36.
Segundo principio datermodinámica Calquera proceso que ocorre espontaneamente produce un aumento da entropía do universo Criterio de espontaneidade: S univ > 0 Δ S universo = Δ S sistema + Δ S arredores > 0
37.
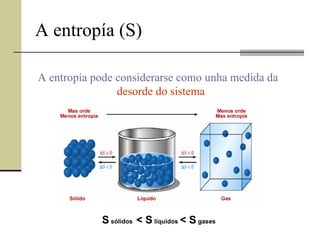
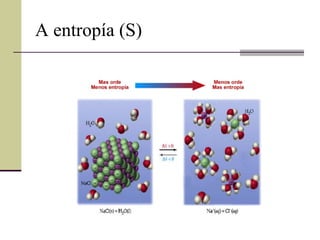
A entropía (S)A entropía pode considerarse como unha medida da desorde do sistema S sólidos < S líquidos < S gases
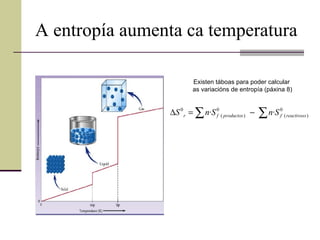
A entropía aumentaca temperatura Existen táboas para poder calcular as variacións de entropía (páxina 8)
40.
A enerxía librede GIBBS ( Δ G ) Como xa vimos a determinación da espontaneidade dunha reacción pode determinarse se coñecemos a variación da entropía total (sistema + arredores) Δ S UNIVERSO > 0 O americano J. W. Gibbs estableceu unha nova magnitude para determinar a espontaneidade dunha reacción: A enerxía libre de Gibbs ( Δ G) : Máxima cantidade de traballo que pode converterse en traballo útil Para o cálculo da enerxía libre poderemos empregar a expresión anterior ou táboas como as da páxina 9
41.
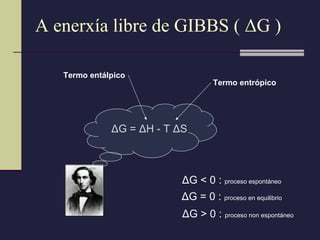
A enerxía librede GIBBS ( Δ G ) Δ G = Δ H - T Δ S Δ G < 0 : proceso espontáneo Δ G = 0 : proceso en equilibrio Δ G > 0 : proceso non espontáneo Termo entálpico Termo entrópico
42.
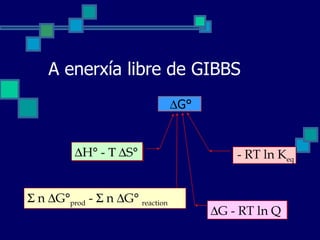
A enerxía librede GIBBS G° n G° prod - n G° reaction H° - S° - RT ln K eq G - RT ln Q