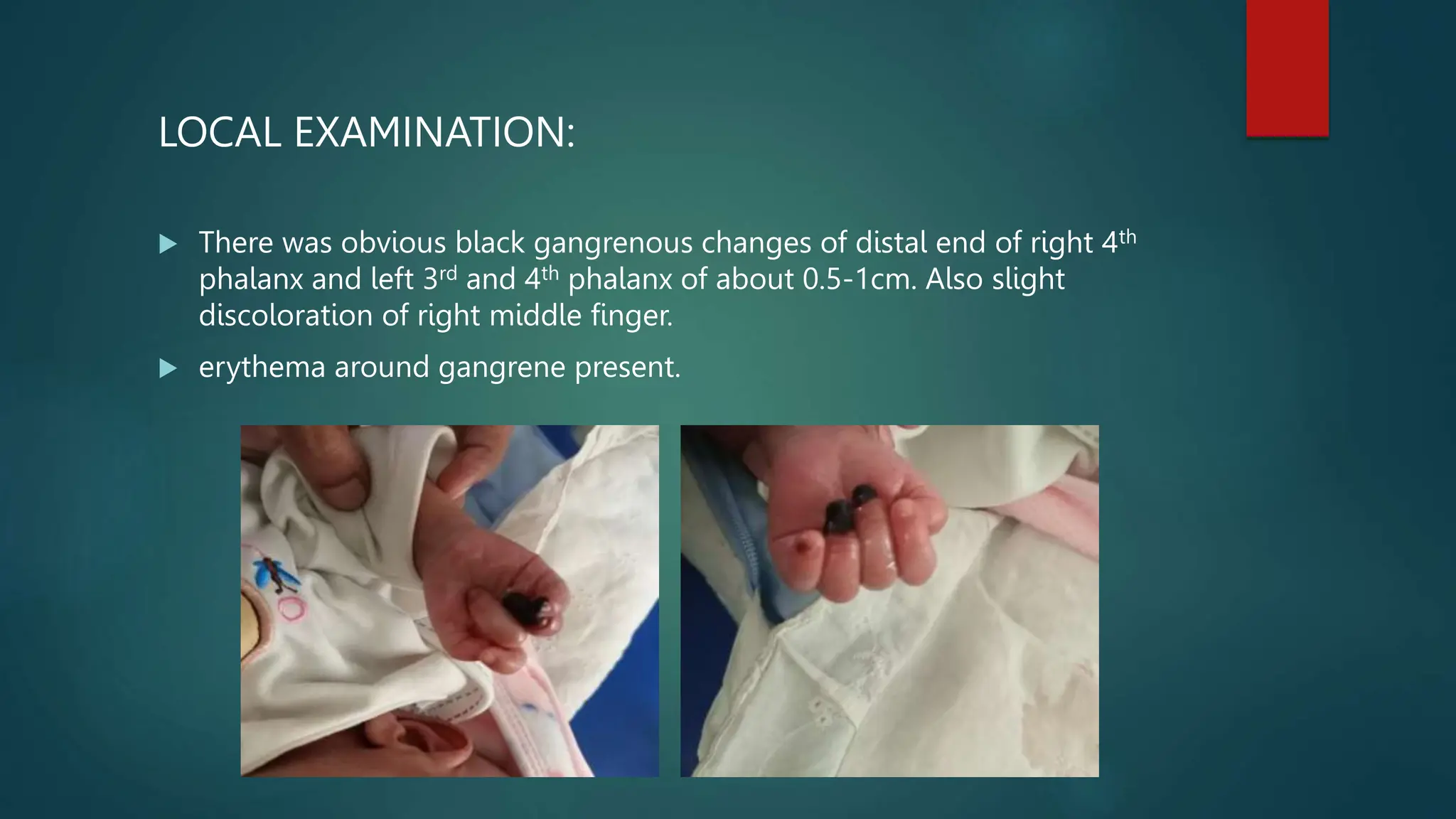
The document details the case presentation of a female infant born on February 26, 2023, who developed acute gastroenteritis and exhibited signs of gangrene in the fingers. Initial treatment at a local clinic with antibiotics was ineffective, leading to admission to the NICU where she was diagnosed with protein S deficiency causing thromboembolic phenomena. The infant received supportive care, including oxygen therapy and anticoagulation treatment, resulting in improvement and subsequent discharge with follow-up instructions.
























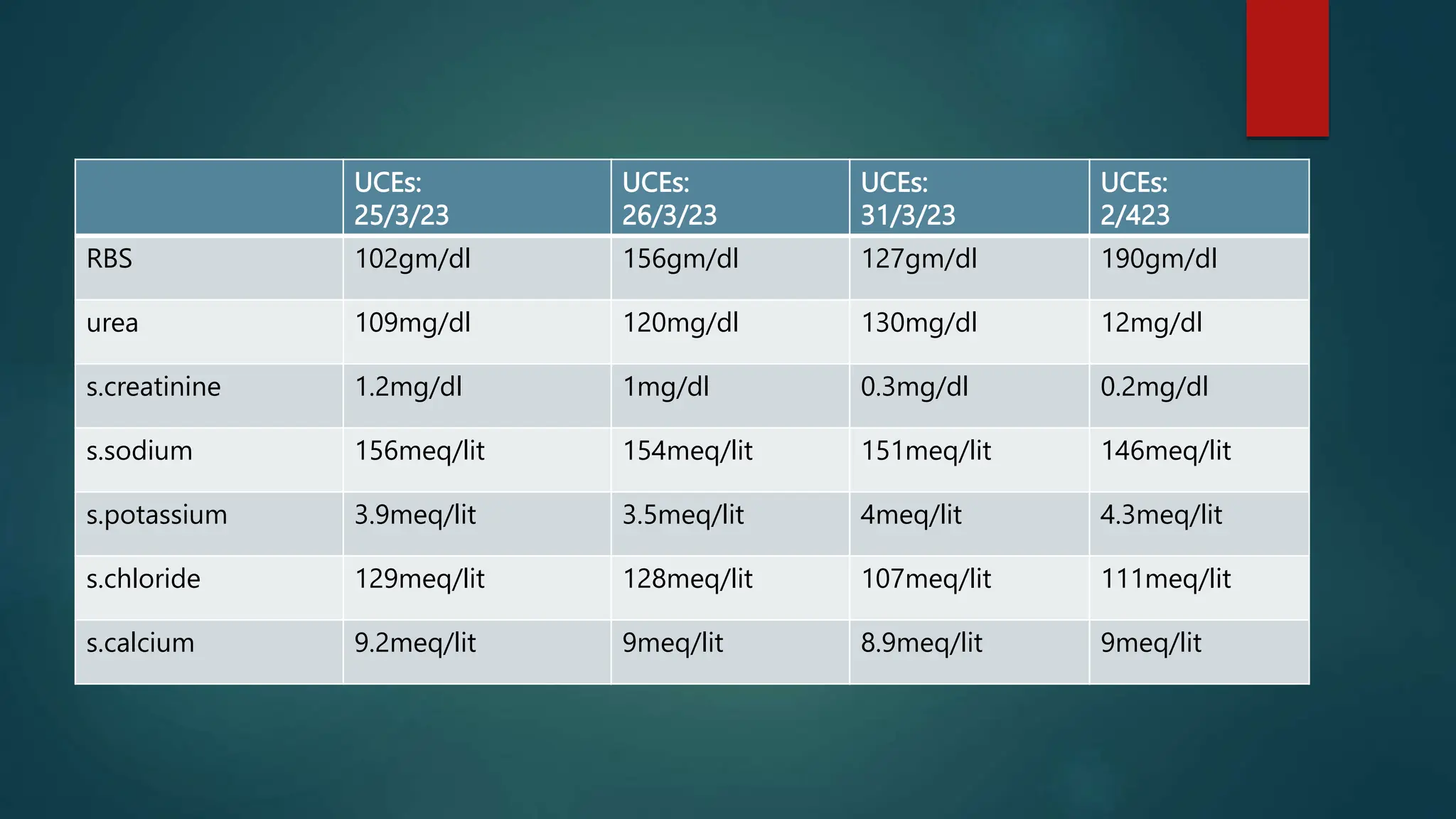

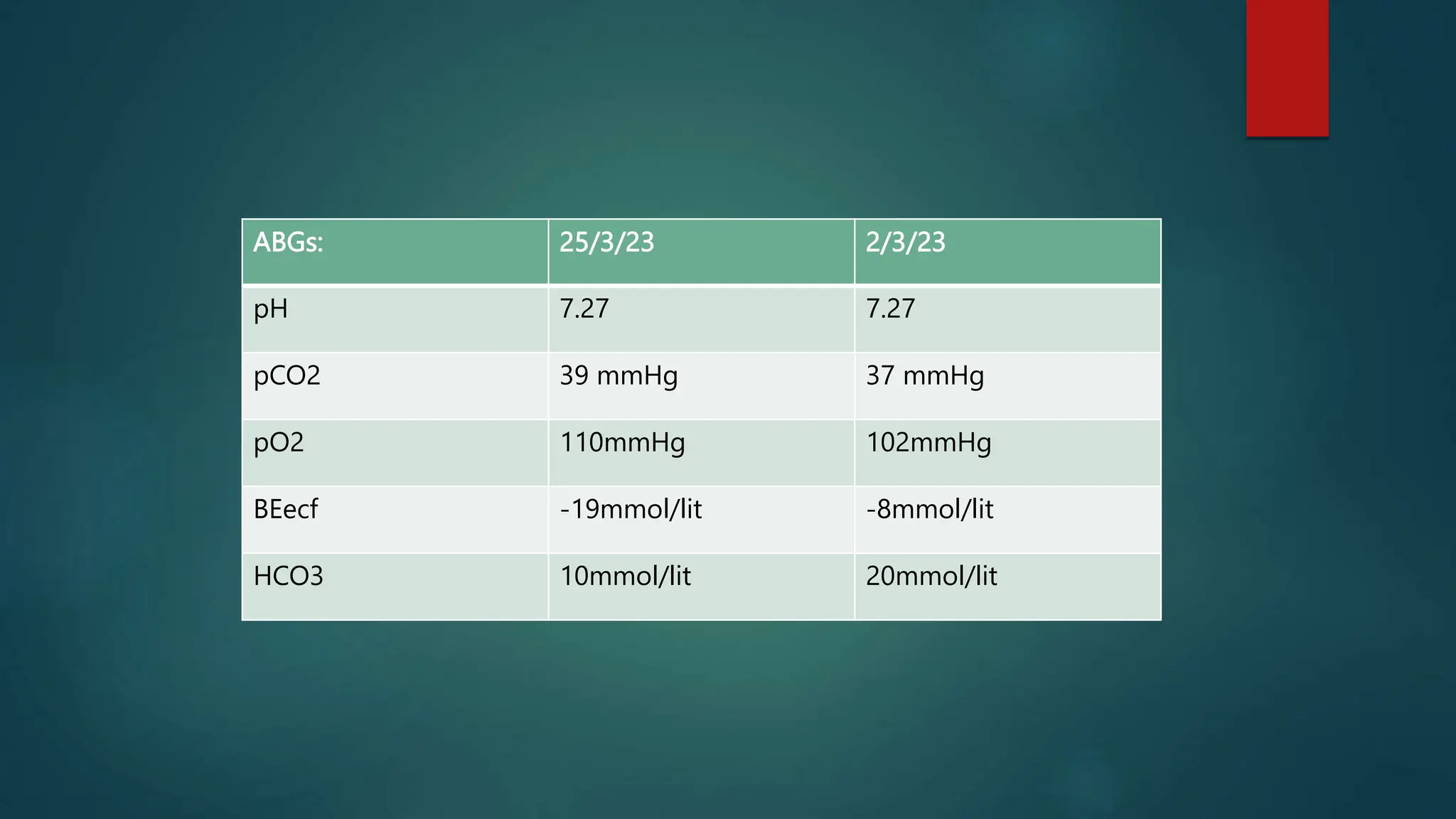
















![Laboratory Investigations:
Choice and interpretation of protein S assay — Protein S deficiency is the most
difficult of the hereditary thrombophilias to document with certainty. As
previously noted, protein S levels in the general population vary more widely
than those of protein C or antithrombin.
Free protein S is our preferred approach to screening as it appears to be the
best test for true deficiency [72,73]. Combining free protein S levels with other
testing such as a functional assay does not appear to improve diagnostic
accuracy.](https://image.slidesharecdn.com/an-240724033923-cc6ed827/75/Protein-C-deficiency-in-Neonate-rare-presentation-45-2048.jpg)




















































