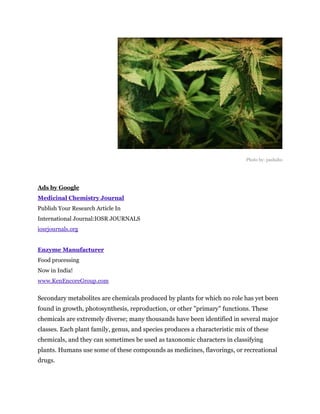
Secondary metabolites are chemicals produced by plants that are not essential for primary functions like growth, photosynthesis or reproduction. They include a wide variety of compounds classified by chemical structure or pathway. Many secondary metabolites help defend plants from herbivores and pathogens. Some have uses as medicines, flavors or recreational drugs and influence plant interactions with other organisms.



![Jump to: navigation, search
Cellulose[1]
Identifiers
CAS number 9004-34-6
UNII SMD1X3XO9M
EC-number 232-674-9
ChEMBL CHEMBL1201676
Properties
Molecular formula (C6H10O5)n
Appearance white powder
Density 1.5 g/cm3
Melting point decomp.
Solubility in water none](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-9-320.jpg)
![Hazards
EU Index not listed
NFPA 704 1
1
0
Related compounds
Related compounds Starch
(verify) (what is: / ?)
Except where noted otherwise, data are given for materials
in their standard state (at 25 °C, 100 kPa)
Infobox references
Cellulose is an organic compound with the formula (C6H10O5)n, a polysaccharide consisting of a
linear chain of several hundred to over ten thousand β(1→4) linked D-glucose units.[2][3]
Cellulose is the structural component of the primary cell wall of green plants, many forms of
algae and the oomycetes. Some species of bacteria secrete it to form biofilms. Cellulose is the
most common organic compound on Earth. About 33% of all plant matter is cellulose (the
cellulose content of cotton fiber is 90%, that of wood is 40–50% and that of dried hemp is
approximately 75%).[4][5][6]
For industrial use, cellulose today is mainly obtained from wood pulp and cotton. Cellulose is
mainly used to produce paperboard and paper; to a smaller extent it is converted into a wide
variety of derivative products such as cellophane and rayon. Converting cellulose from energy
crops into biofuels such as cellulosic ethanol is under investigation as an alternative fuel source.
Some animals, particularly ruminants and termites, can digest cellulose with the help of
symbiotic micro-organisms that live in their guts. Humans can digest cellulose to some](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-10-320.jpg)
![extent,[7][8] however it mainly acts as a hydrophilic bulking agent for feces and is often referred
to as "dietary fiber".
Contents
1 History
2 Products
o 2.1 Cellulose source and energy crops
3 Structure and properties
4 Assaying a cellulose-containing material
5 Biosynthesis
6 Breakdown (cellulolysis)
7 Hemicellulose
8 Derivatives
9 References
10 External links
[edit] History
Cellulose was discovered in 1838 by the French chemist Anselme Payen, who isolated it from
plant matter and determined its chemical formula.[2][9][10] Cellulose was used to produce the first
successful thermoplastic polymer, celluloid, by Hyatt Manufacturing Company in 1870.
Hermann Staudinger determined the polymer structure of cellulose in 1920. The compound was
first chemically synthesized (without the use of any biologically derived enzymes) in 1992, by
Kobayashi and Shoda.[11]
Cellulose in a plant cell.
[edit] Products](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-11-320.jpg)
![Cotton fibres represent the purest natural form of cellulose, containing more than 90% of this
carbohydrate.
See also: dissolving pulp and pulp (paper)
The kraft process is used to separate cellulose from lignin, another major component of plant
matter. Cellulose is the major constituent of paper, paperboard, and card stock and of textiles
made from cotton, linen, and other plant fibers.
Cellulose can be converted into cellophane, a thin transparent film, and into rayon, an important
fiber that has been used for textiles since the beginning of the 20th century. Both cellophane and
rayon are known as "regenerated cellulose fibers"; they are identical to cellulose in chemical
structure and are usually made from dissolving pulp via viscose. A more recent and
environmentally friendly method to produce a form of rayon is the Lyocell process. Cellulose is
the raw material in the manufacture of nitrocellulose (cellulose nitrate) which is used in
smokeless gunpowder and as the base material for celluloid used for photographic and movie
films until the mid 1930s.
Cellulose is used to make water-soluble adhesives and binders such as methyl cellulose and
carboxymethyl cellulose which are used in wallpaper paste. Microcrystalline cellulose (E460i)
and powdered cellulose (E460ii) are used as inactive fillers in tablets[12] and as thickeners and
stabilizers in processed foods. Cellulose powder is for example used in Kraft's Parmesan cheese
to prevent caking inside the tube.
Cellulose is used in the laboratory as the stationary phase for thin layer chromatography.
Cellulose fibers are also used in liquid filtration, sometimes in combination with diatomaceous
earth or other filtration media, to create a filter bed of inert material. Cellulose is further used to
make hydrophilic and highly absorbent sponges.
Cellulose insulation made from recycled paper is becoming popular as an environmentally
preferable material for building insulation. It can be treated with boric acid as a fire retardant.
Cellulose consists of crystalline and amorphous regions. By treating it with strong acid, the
amorphous regions can be broken up, thereby producing nanocrystalline cellulose, a novel
material with many desirable properties.[13] Recently, nanocrystalline cellulose was used as the](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-12-320.jpg)
![filler phase in bio-based polymer matrices to produce nanocomposites with superior thermal and
mechanical properties. [14]
[edit] Cellulose source and energy crops
Main article: Energy crop
The major combustible component of non-food energy crops is cellulose, with lignin second.
Non-food energy crops are more efficient than edible energy crops (which have a large starch
component), but still compete with food crops for agricultural land and water resources.[15]
Typical non-food energy crops include industrial hemp (though outlawed in some countries),
switchgrass, Miscanthus, Salix (willow), and Populus (poplar) species.
Some bacteria can convert cellulose into ethanol which can then be used as a fuel; see cellulosic
ethanol.
A strand of cellulose (conformation Iα), showing the hydrogen bonds (dashed) within and between
cellulose molecules.
[edit] Structure and properties
Cellulose has no taste, is odorless, is hydrophilic with the contact angle of 20–30,[16] is insoluble
in water and most organic solvents, is chiral and is biodegradable. It can be broken down
chemically into its glucose units by treating it with concentrated acids at high temperature.
Cellulose is derived from D-glucose units, which condense through β(1→4)-glycosidic bonds.
This linkage motif contrasts with that for α(1→4)-glycosidic bonds present in starch, glycogen,
and other carbohydrates. Cellulose is a straight chain polymer: unlike starch, no coiling or
branching occurs, and the molecule adopts an extended and rather stiff rod-like conformation,
aided by the equatorial conformation of the glucose residues. The multiple hydroxyl groups on
the glucose from one chain form hydrogen bonds with oxygen atoms on the same or on a
neighbor chain, holding the chains firmly together side-by-side and forming microfibrils with](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-13-320.jpg)
![high tensile strength. This strength is important in cell walls, where the microfibrils are meshed
into a carbohydrate matrix, conferring rigidity to plant cells.
A triple strand of cellulose showing the hydrogen bonds (cyan lines) between glucose strands
Compared to starch, cellulose is also much more crystalline. Whereas starch undergoes a
crystalline to amorphous transition when heated beyond 60–70 °C in water (as in cooking),
cellulose requires a temperature of 320 °C and pressure of 25 MPa to become amorphous in
water.[17]
Several different crystalline structures of cellulose are known, corresponding to the location of
hydrogen bonds between and within strands. Natural cellulose is cellulose I, with structures Iα
and Iβ. Cellulose produced by bacteria and algae is enriched in Iα while cellulose of higher plants
consists mainly of Iβ. Cellulose in regenerated cellulose fibers is cellulose II. The conversion of
cellulose I to cellulose II is irreversible, suggesting that cellulose I is metastable and cellulose II
is stable. With various chemical treatments it is possible to produce the structures cellulose III
and cellulose IV.[18]
Many properties of cellulose depend on its chain length or degree of polymerization, the number
of glucose units that make up one polymer molecule. Cellulose from wood pulp has typical chain
lengths between 300 and 1700 units; cotton and other plant fibers as well as bacterial cellulose
have chain lengths ranging from 800 to 10,000 units.[11] Molecules with very small chain length
resulting from the breakdown of cellulose are known as cellodextrins; in contrast to long-chain
cellulose, cellodextrins are typically soluble in water and organic solvents.
Plant-derived cellulose is usually found in a mixture with hemicellulose, lignin, pectin and other
substances, while microbial cellulose is quite pure, has a much higher water content, and consists
of long chains.
Cellulose is soluble in cupriethylenediamine (CED), cadmiumethylenediamine (Cadoxen), N-
methylmorpholine N-oxide and lithium chloride / dimethylformamide.[19] This is used in the
production of regenerated celluloses (such as viscose and cellophane) from dissolving pulp.
[edit] Assaying a cellulose-containing material
Given a cellulose-containing material, the carbohydrate portion that does not dissolve in a 17.5%
solution of sodium hydroxide at 20 °C is α cellulose, which is true cellulose. Acidification of the
extract precipitates β cellulose. The portion that dissolves in base but does not precipitate with
acid is γ cellulose.](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-14-320.jpg)
![Cellulose can be assayed using a method described by Updegraff in 1969, where the fiber is
dissolved in acetic and nitric acid to remove lignin, hemicellulose, and xylosans. The resulting
cellulose is allowed to react with anthrone in sulfuric acid. The resulting coloured compound is
assayed spectrophotometrically at a wavelength of approximately 635 nm.
In addition, cellulose is represented by the difference between acid detergent fiber (ADF) and
acid detergent lignin (ADL).
[edit] Biosynthesis
Location and arrangement of cellulose microfibrils in the plant cell wall
In vascular plants cellulose is synthesized at the plasma membrane by rosette terminal complexes
(RTCs). The RTCs are hexameric protein structures, approximately 25 nm in diameter, that
contain the cellulose synthase enzymes that synthesise the individual cellulose chains.[20] Each
RTC floats in the cell's plasma membrane and "spins" a microfibril into the cell wall.
RTCs contain at least three different cellulose synthases, encoded by CesA genes, in an unknown
stoichiometry.[21] Separate sets of CesA genes are involved in primary and secondary cell wall
biosynthesis.
Cellulose synthesis requires chain initiation and elongation, and the two processes are separate.
CesA glucosyltransferase initiates cellulose polymerization using a steroid primer, sitosterol-
beta-glucoside, and UDP-glucose.[22] Cellulose synthase utilizes UDP-D-glucose precursors to
elongate the growing cellulose chain. A cellulase may function to cleave the primer from the
mature chain.
Cellulose is also synthesised by animals, particularly in the tests of ascidians (where the cellulose
was historically termed "tunicine") although it is also a minor component of mammalian
connective tissue.[23]
[edit] Breakdown (cellulolysis)
Cellulolysis is the process of breaking down cellulose into smaller polysaccharides called
cellodextrins or completely into glucose units; this is a hydrolysis reaction. Because cellulose
molecules bind strongly to each other, cellulolysis is relatively difficult compared to the
breakdown of other polysaccharides.[24] Processes do exist however for the breakdown of](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-15-320.jpg)
![cellulose such as the Lyocell process [25] which uses a combination of heated water and acetone
to break down the cellulose strands.
Most mammals have only very limited ability to digest dietary fibres such as cellulose. Some
ruminants like cows and sheep contain certain symbiotic anaerobic bacteria (like Cellulomonas)
in the flora of the rumen, and these bacteria produce enzymes called cellulases that help the
microorganism to break down cellulose; the breakdown products are then used by the bacteria
for proliferation. The bacterial mass is later digested by the ruminant in its digestive system
(stomach and small intestine). Similarly, lower termites contain in their hindguts certain
flagellate protozoa which produce such enzymes; higher termites contain bacteria for the job.
Some termites may also produce cellulase of their own.[26] Fungi, which in nature are responsible
for recycling of nutrients, are also able to break down cellulose.
The enzymes utilized to cleave the glycosidic linkage in cellulose are glycoside hydrolases
including endo-acting cellulases and exo-acting glucosidases. Such enzymes are usually secreted
as part of multienzyme complexes that may include dockerins and carbohydrate-binding
modules.[27]
[edit] Hemicellulose
Main article: Hemicellulose
Hemicellulose is a polysaccharide related to cellulose that comprises about 20% of the biomass
of most plants. In contrast to cellulose, hemicellulose is derived from several sugars in addition
to glucose, especially xylose but also including mannose, galactose, rhamnose, and arabinose.
Hemicellulose consists of shorter chains – around 200 sugar units. Furthermore, hemicellulose is
branched, whereas cellulose is unbranched.
[edit] Derivatives
The hydroxyl groups (-OH) of cellulose can be partially or fully reacted with various reagents to
afford derivatives with useful properties like mainly cellulose esters and cellulose ethers (-OR).
In principle, though not always in current industrial practice, cellulosic polymers are renewable
resources.
Ester derivatives include:
Cellulose
Reagent Example Reagent Group R
ester
Organic Organic Acetic acid and acetic
Cellulose acetate H or -(C=O)CH3
esters acids anhydride
Acetic acid and acetic
Cellulose triacetate -(C=O)CH3
anhydride](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-16-320.jpg)

![chloroethane
Cold/ho Gelling and
Hydroxyalk Hydroxyethyl Ethylene
Epoxides -CH2CH2OH t water thickening
yl cellulose oxide
soluble agent
Hydroxypropy - Cold
Propylene
l cellulose CH2CH(OH)C water E463
oxide
(HPC) H3 soluble
Chloromethan
Hydroxyethyl Cold Production of
e and -CH3 or -
methyl water cellulose
ethylene CH2CH2OH
cellulose soluble films
oxide
Viscosity
Hydroxypropy Chloromethan modifier,
-CH3 or - Cold
l methyl e and gelling,
CH2CH(OH)C water E464
cellulose propylene foaming and
H3 soluble
(HPMC) oxide binding
agent
Ethyl Chloroethane
-CH2CH3 or—
hydroxyethyl and ethylene E467
CH2CH2OH
cellulose oxide
Often used
as its sodium
Carboxymethy Cold/Ho
Carboxyalk Halogenated Chloroacetic salt, sodium
l cellulose -CH2COOH t water E466
yl carboxylic acids acid carboxymeth
(CMC) soluble
yl cellulose
(NaCMC)
The sodium carboxymethyl cellulose can be cross-linked to give the croscarmellose sodium
(E468) for use as a disintegrant in pharmaceutical formulations.
[edit] References
1. ^ Nishiyama, Yoshiharu; Langan, Paul; Chanzy, Henri (2002). "Crystal Structure and Hydrogen-Bonding
System in Cellulose Iβ from Synchrotron X-ray and Neutron Fiber Diffraction". J. Am. Chem. Soc 124 (31):
9074–82. doi:10.1021/ja0257319. PMID 12149011..
ab
2. ^ Crawford, R. L. (1981). Lignin biodegradation and transformation. New York: John Wiley and Sons.
ISBN 0-471-05743-6.](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-18-320.jpg)
![3. ^ Updegraff DM (1969). "Semimicro determination of cellulose in biological materials". Analytical
Biochemistry 32 (3): 420–424. doi:10.1016/S0003-2697(69)80009-6. PMID 5361396.
4. ^ Cellulose. (2008). In Encyclopædia Britannica. Retrieved January 11, 2008, from Encyclopædia Britannica
Online.
5. ^ Chemical Composition of Wood
6. ^ G. Buschle-Diller, C. Fanter, F. Loth (April 1999). "Structural changes in hemp fibers as a result of
enzymatic hydrolysis with mixed enzyme systems". Textile Research Journal 69 (4): 244-251.
http://www.globalhemp.com/1999/04/structural-changes-in-hemp-fibers-as-a-result-of-enzymatic-
hydrolysis-with-mixed-enzyme-systems.html.
7. ^ Slavin, JL; Brauer, PM; Marlett, JA (1981). "Neutral detergent fiber, hemicellulose and cellulose
digestibility in human subjects.". The Journal of Nutrition 111 (2): 287–97. PMID 6257867.
8. ^ Joshi, S; Agte, V (1995). "Digestibility of dietary fiber components in vegetarian men.". Plant foods for
human nutrition (Dordrecht, Netherlands) 48 (1): 39–44. doi:10.1007/BF01089198. PMID 8719737.
9. ^ A. Payen (1838) "Mémoire sur la composition du tissu propre des plantes et du ligneux" (Memoir on the
composition of the tissue of plants and of woody [material]), Comptes rendus, vol. 7, pages 1052-1056.
Payen added appendices to this paper on December 24, 1838 (see: Comptes rendus, vol. 8, page 169
(1839)) and on February 4, 1839 (see: Comptes rendus, vol. 9, page 149 (1839)). A committee of the
French Academy of Sciences reviewed Payen's findings in : Jean-Baptiste Dumas (1839) "Rapport sur un
mémoire de M. Payen, relatif à la composition de la matière ligneuse" (Report on a memoir of Mr. Payen,
regarding the composition of woody matter), Comptes rendus, vol. 8, pages 51-53. In this report, the word
"cellulose" is coined and author points out the similarity between the empirical formula of cellulose and
that of "dextrine" (starch). The above articles are reprinted in: Brongniart and Guillemin, eds., Annales des
sciences naturelles ..., 2nd series, vol. 11 (Paris, France: Crochard et Cie., 1839), pages 21-31.
10. ^ Young, Raymond (1986). Cellulose structure modification and hydrolysis. New York: Wiley. ISBN 0-471-
82761-4.
ab
11. ^ Klemm, Dieter; Brigitte Heublein, Hans-Peter Fink, Andreas Bohn (2005). "Cellulose: Fascinating
Biopolymer and Sustainable Raw Material". ChemInform 36 (36). doi:10.1002/chin.200536238.
12. ^ Weiner, Myra L.; Lois A. Kotkoskie (2000). Excipient Toxicity and Safety. New York ; Dekker. p. 210.
ISBN 0-8247-8210-0.
13. ^ Peng, B. L., Dhar, N., Liu, H. L. and Tam, K. C. (2011). "Chemistry and applications of nanocrystalline
cellulose and its derivatives: A nanotechnology perspective.". The Canadian Journal of Chemical
Engineering 89 (5): 1191–1206.
http://www.arboranano.ca/pdfs/Chemistry%20and%20applications%20of%20nanocrystalline%20cellulos
e%20and%20its%20derivatives%20A%20nanotechnology%20perspective-2011.pdf.
14. ^ Lawrence Pranger and Rina Tannenbaum "Biobased nanocomposites prepared by in situ polymerization
of furfuryl alcohol with cellulose whiskers or montmorillonite clay" Macromolecules 41 (2008) 8682.
http://dx.doi.org/10.1021/ma8020213
15. ^ Holt-Gimenez, Eric 2007. Biofuels: Myths of the Agrofuels Transition. Backgrounder. Institute for Food
and Development Policy, Oakland, CA. 13:2
16. ^ Charles A. Bishop, ed. (2007). Vacuum deposition onto webs, films, and foils, Volume 0, Issue 8155.
p. 165. ISBN 0-8155-1535-9. http://books.google.com/books?id=vP9E3z7o6iIC&pg=PA165.
17. ^ Deguchi, Shigeru; Tsujii, Kaoru; Horikoshi, Koki (2006). "Cooking cellulose in hot and compressed water".
Chemical Communications (31): 3293. doi:10.1039/b605812d.
18. ^ Structure and morphology of cellulose by Serge Pérez and William Mackie, CERMAV-CNRS, 2001.
Chapter IV.
19. ^ Stenius, Per (2000). "1". Forest Products Chemistry. Papermaking Science and Technology. 3. Finland:
Fapet OY. p. 35. ISBN 952-5216-03-9.
20. ^ Kimura, S; Laosinchai, W; Itoh, T; Cui, X; Linder, CR; Brown Jr, RM (1999). "Immunogold labeling of
rosette terminal cellulose-synthesizing complexes in the vascular plant vigna angularis". The Plant cell 11
(11): 2075–86. doi:10.2307/3871010. JSTOR 3871010. PMC 144118. PMID 10559435.
//www.ncbi.nlm.nih.gov/pmc/articles/PMC144118/.
21. ^ Taylor, N. G. (2003). "Interactions among three distinct CesA proteins essential for cellulose synthesis".
Proceedings of the National Academy of Sciences 100 (3): 1450. doi:10.1073/pnas.0337628100.](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-19-320.jpg)
![22. ^ Peng, L; Kawagoe, Y; Hogan, P; Delmer, D (2002). "Sitosterol-beta-glucoside as primer for cellulose
synthesis in plants". Science 295 (5552): 147–50. doi:10.1126/science.1064281. PMID 11778054.
23. ^ Endean, The Test of the Ascidian, Phallusia mammillata, Quarterly Journal of Microscopical Science, Vol.
102, part 1, pp. 107-117, 1961.
24. ^ David G. Barkalow, Roy L. Whistler, "Cellulose", in AccessScience, McGraw-Hill, doi:10.1036/1097-
8542.118200. Retrieved 11 January 2008.
25. ^ H.Lyocell, "Cellulose" Issue 41, pp 419
26. ^ Tokuda, G; Watanabe, H (22 June 2007). "Hidden cellulases in termites: revision of an old hypothesis".
Biology Letters 3 (3): 336–339. doi:10.1098/rsbl.2007.0073. PMC 2464699. PMID 17374589.
http://rsbl.royalsocietypublishing.org/content/3/3/336.long
27. ^ Brás, Natércia; N. M. F. S. A. Cerqueira, P. A. Fernandes, M. J. Ramos (2008). "Carbohydrate Binding
Modules from family 11: Understanding the binding mode of polysaccharides". International Journal of
Quantum Chemistry 108 (11): 2030–2040. doi:10.1002/qua.21755.
[edit] External links
Structure and morphology of cellulose by Serge Pérez and William Mackie, CERMAV-CNRS
Cellulose, by Martin Chaplin, London South Bank University
Clear description of a cellulose assay method at the Cotton Fiber Biosciences unit of the USDA.
Cellulose films could provide flapping wings and cheap artificial muscles for robots -
TechnologyReview.com
Using cellulase enzymes in the bioethanol process
A list of cellulolytic bacteria
v
t
e
Types of carbohydrates
Aldose
Furanose
General
Ketose
Pyranose
Anomer
Geometry Cyclohexane conformation
Mutarotation](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-20-320.jpg)















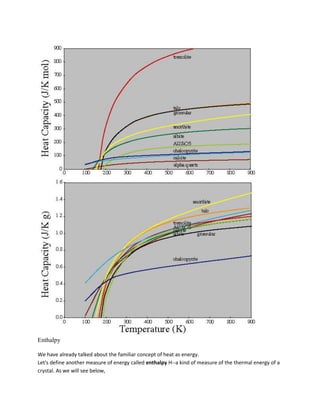





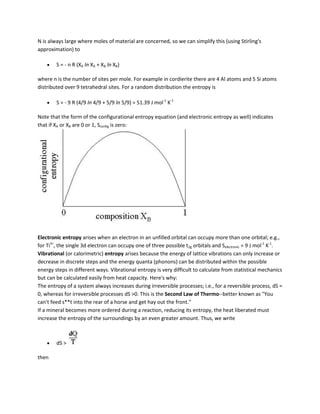











![A = (Vmix - XB B) / XA
Entropy of Mixing
The entropy of mixing is never zero because mixing increases entropy. As we discussed days ago, the
entropy of mixing (i.e., the configurational entropy) is
Smix = -R (Xi ln Xi)
where i = 1..n is the number of sites over which mixing is occurring.
Enthalpy of Mixing
Enthalpies also do not combined ideally (linearly) in mixtures because the mixture may have stronger
bonds than were present in either of the unmixed phases. The excess enthalpy is
Hmix = 0.5 * N z XAXB [2 AB - AA - BB]
where AB is the interaction energy among A-B atoms, AA is the interaction energy among A-A atoms,
and BB is the interaction energy among B-B atoms.
Gibbs Free Energy of Mixing
Recall that all spontaneous processes/reactions occur because of a decrease in Gibbs free energy. It
should therefore not surprise you that the Gibbs free energy of mixing is always negative--otherwise
mixing would not occur. The fact that A < G°A and B < G°B illustrates why compounds combine
spontaneously--each compound is able to lower its free energy.](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-60-320.jpg)
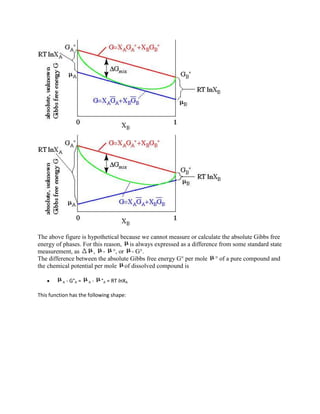




![is the law of mass action (which actually discusses the action of chemical potential rather than mass).
We can also write for 298 K and 1 atm
rH° -T rS° = - RT ln K
and for any P and T of interest:
rH1,Tref + CPdT - T rSTref + dT + rVP = - RT ln K
This has been called "the most important equation in thermodynamics," so you'd better like it(!) The
equilibrium constant K is a function of 1/T
-ln K = ( rG° / RT) = [( rH / RT) - ( rS / R)]
Which looks like
Activity Models (Activity-Composition Relations) for Crystalline Solutions
Garnets are solid solutions of
component abbrev. Formula
pyrope prp Mg3Al2Si3O12
almandine alm Fe3Al2Si3O12
grossular grs Ca3Al2Si3O12
spessartine sps Mn3Al2Si3O12
andradite and Ca3Fe23+Si3O12
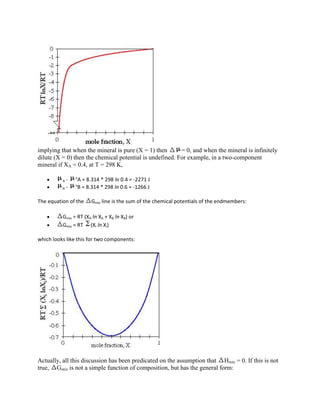
Mixing models derive from entropy considerations. In particular the relation](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-67-320.jpg)
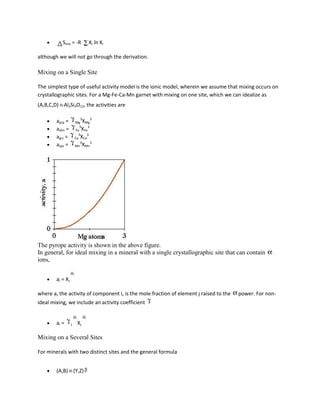
![Fe3Al2Si3O12 + KMg3AlSi3O10(OH)2 = Mg3Al2Si3O12 + KFe3AlSi3O10(OH)2
almandine + phlogopite = pyrope + annite
We can reduce this reaction to a simple exchange vector:
(FeMg)gar+1 = (FeMg)bio-1
Popular thermometers include garnet-biotite (GARB), garnet-clinopyroxene, garnet-hornblende, and
clinopyroxene-orthopyroxene; all of these are based on the exchange of Fe and Mg, and are excellent
thermometers because rV is small, such that
=
is large (i.e., the reactions have steep slopes and are little influenced by pressure). Let's write the
equilibrium constant for the GARB exchange reaction
K = (aprpaann)/(aalmaphl)
thus
rG = -RT ln (aprpaann)/(aalmaphl)
This equation implies that the activities of the Fe and Mg components of biotite and garnet are a
function of Gibbs free energy change and thus are functions of pressure and temperature.
If we assume ideal behavior ( = 1) in garnet and biotite and assume that there is mixing on only 1 site
aalm = Xalm3 = [Fe/(Fe + Mg + Ca + Mn)]3
aprp = Xprp3 = [Mg/(Fe + Mg)]3
aann = Xann3 = [Fe/(Fe + Mg)]3
aphl = Xphl3 = [Mg/(Fe + Mg)]3
Thus the equilibrium constant is
K = (XMggar XFebio)/(XFegar XMgbio)
When discussing element partitioning it is common to define a distribution coefficient KD, which is just
the equilibrium constant without the exponent (this just describes the partitioning of elements and not
the partitioning of chemical potential):
KD = (XMggar XFebio)/(XFegar XMgbio) = (Mg/Fe)gar /(Mg/Fe)bio = K1/3](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-70-320.jpg)
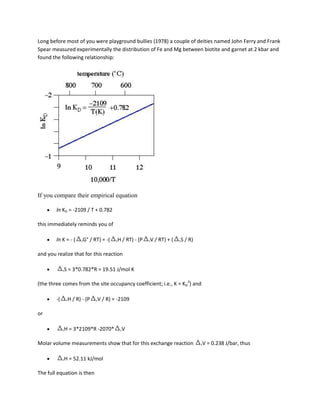
![52,110 - 19.51*T(K) + 0.238*P(bar) + 3RT ln KD = 0
To plot the KD lines in PT space
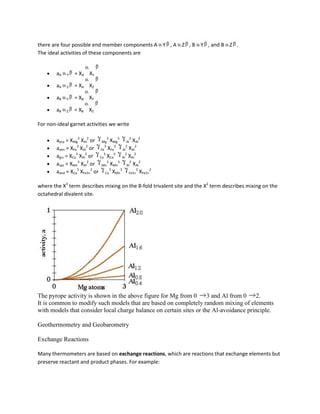
Net-Transfer Reactions
Net-transfer reactions are those that cause phases to appear or disappear. Geobarometers are often
based on net-transfer reactions because rV is large and relatively insensitive to temperature. A
popular one is GASP:
3CaAl2Si2O8 = Ca3Al2Si3O12 + 2Al2SiO5 + SiO2
anorthite = grossular + kyanite + quartz
which describes the high-pressure breakdown of anorthite.
For this reaction
rG = -RT ln [(aqtzaky2agrs) / aan3] = -RT ln agrs / aan3
(the activities of quartz and kyanite are one because they are pure phases). A best fit through the
experimental data for this reaction by Andrea Koziol and Bob Newton yields
P(bar) = 22.80 T(K) - 7317
for rV = -6.608 J/bar. Again, if we use](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-72-320.jpg)
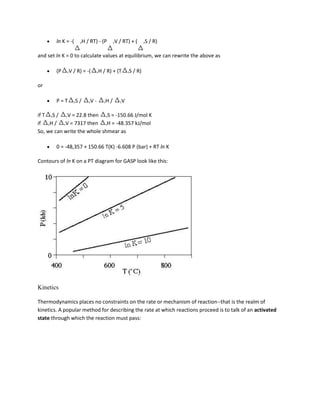
![This change from increasing to decreasing nucleation free energy happens because the
surface:volume ratio decreases with increasing radius, so the negative volume free energy term
that favors nucleation eventually overwhelms the positive surface and strain free energy terms
that oppose nucleation. We can re-order the terms in the above equation to calculate the critical
size of the nucleus rc
rc = -2 /( VG + )
If, at the critical radius, nG = G*, the activation energy for nucleation is
3
G* = 16 / 3( VG + )2
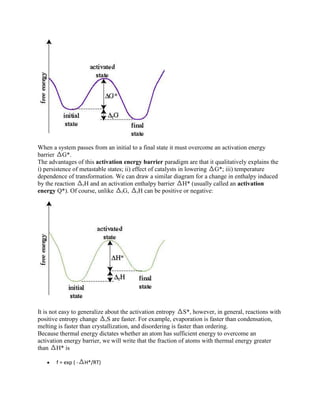
Growth
Following nucleation, grains of the product phase(s) grow to replace the parent phase(s). Either the
reaction is polymorphic, or atoms of the parent phase must dissolve, be transported, and then attach
themselves to the product phase. The rate of growth is thus controlled by the rate of the slowest of
these three steps, and is described as either interface controlled or diffusion controlled.
Turnbull's (1956) formulation to quantify interface-controlled growth rate is:
= (kT/h) exp( -Q*/RT)[( - rG/RT)]
where is an "interface jump distance" and the rest you will recognize from previous equations. The [( -
rG/RT)] term indicates that the rate of growth depends on the Gibbs free energy change of reaction.
As rG 0, this term also approaches zero. As rG , this term approaches 1.
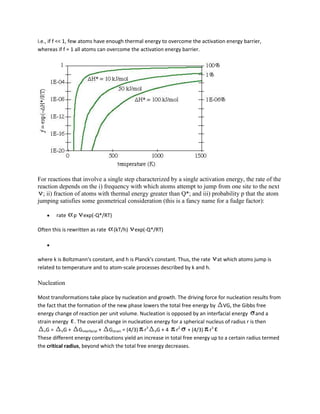
Transformation: Nucleation + Growth](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-76-320.jpg)
![A complete transformation involves nucleation and growth--the product phases must form and grow
and the parent phases must be consumed. The letter _ is often used to indicate 'reaction progress' or
'degree of transformation,' and varies from 0 (no reaction) to 1 (complete reaction). The transformation
is the time-integrated result of ongoing nucleation and growth, which can vary in time and space:
= 1 - exp[(4/3) dt]
Just as an example, the general rate equation for interface-controlled (i.e., not diffusion-controlled)
growth of nuclei formed on grain boundaries is
2
= 1 - exp{(6.7/d) [1-exp( [ (t - )2 - y2] d )] dy}
In their simplest form, these equations have the form
= 1 - exp(-ktn)
Flip this around to
exp(-ktn) = 1 -
linearize by means of logarithms
-ktn = ln (1 - )
change signs and cleverly insert ln 1
ktn = ln 1 - ln (1 - )
rewrite the two logarithms as one, recalling that ln (a/b) = ln a - ln b
ktn = ln [1 / (1 - )]
and linearize once more with logarithms to get the final glorious equation
n ln t + ln k = ln [ ln (1 - )]
The Last Supper: A Still Life of Thermodynamics & Kinetics
Zeolites, like laumontite and wairakite, form in oil fields as alteration products of plagioclase and other
Ca-bearing phases. The thermodynamic properties ( H, S, V, CP) of zeolites have been measured, and
we can use those properties to calculate a phase diagram for the CASH system:](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-77-320.jpg)
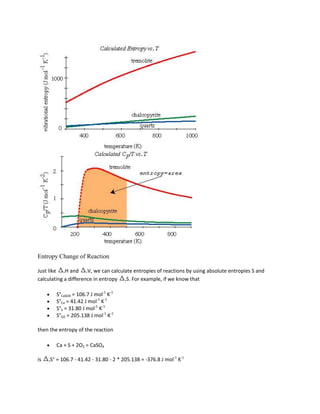
![Laumontite is the phase that is stable at the lowest P and T. Wairakite is the next phase to form, and it
does so via the reaction laumontite _ wairakite + H2O. At higher temperature wairakite + H2O
decompose to anorthite + quartz + H2O.
Kinetic experiments conducted on the reaction laumontite _ wairakite + H2O, using crystals of
laumontite suspended in H2O, reveal the transformation vs. time data in the figure above.
One could use the equation
to interpret the two sub-figures to reveal different n and k values for each temperature. This is the way
rate data were interpreted in the bad old days, but this approach lacks any physical or mechanistic basis.
Instead, we can measure the rate of growth at each temperature and relate the rate of growth to the
Turnbull equation. Above is an example of grain growth data collected for 450°C. The rate of growth is
interpreted to be a constant 2.0 ± 0.3 E-10 m/s. If growth rate data like this can be collected for every
temperature of interest, an activation energy for growth can be calculated:
The activation energy for growth apparently varies from ~196 kJ/mol to ~ 72 kJ/mol depending on the
reaction mechanism. The growth rate data can be combined with similar nucleation rate data to yield a
complete transformation rate equation that can be extrapolated to geologic conditions of interest. The
figure below shows extrapolated growth rates (m/s) for the laumontite _ wairakite + H2O reaction.
search
Potentials
Free energy · Free entropy
Internal energy U(S,V)
Enthalpy H(S,p) = U + pV
Helmholtz free energy A(T,V) = U − TS
Gibbs free energy G(T,p) = H − TS
The four laws of thermodynamics define fundamental physical quantities (temperature, energy, and
entropy) that characterize thermodynamic systems. The laws describe how these quantities behave
under various circumstances, and forbid certain phenomena (such as perpetual motion).
The four laws of thermodynamics are:[1][2][3][4][5][6]](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-78-320.jpg)
![Zeroth law of thermodynamics: If two systems are in thermal equilibrium with a third system,
they must be in thermal equilibrium with each other. This law helps define the notion of
temperature.
First law of thermodynamics: Heat and work are forms of energy transfer. Energy is invariably
conserved but the internal energy of a closed system changes as heat and work are transferred
in or out of it. Equivalently, perpetual motion machines of the first kind are impossible.
Second law of thermodynamics: The entropy of any isolated system not in thermal
equilibrium almost always increases. Isolated systems spontaneously evolve towards thermal
equilibrium—the state of maximum entropy of the system—in a process known as
"thermalization". Equivalently, perpetual motion machines of the second kind are impossible.
Third law of thermodynamics: The entropy of a system approaches a constant value as the
temperature approaches zero. The entropy of a system at absolute zero is typically zero, and in
all cases is determined only by the number of different ground states it has. Specifically, the
entropy of a pure crystalline substance at absolute zero temperature is zero.
Classical thermodynamics describes the exchange of work and heat between systems. It has a special
interest in systems that are individually in states of thermodynamic equilibrium. Thermodynamic
equilibrium is a condition of systems which are adequately described by only macroscopic variables.
Every physical system, however, when microscopically examined, shows apparently random microscopic
statistical fluctuations in its thermodynamic variables of state (entropy, temperature, pressure, etc.).
These microscopic fluctuations are negligible for systems which are nearly in thermodynamic equilibrium
and which are only macroscopically examined. They become important, however, for systems which are
nearly in thermodynamic equilibrium when they are microscopically examined, and, exceptionally, for
macroscopically examined systems that are in critical states,[7] and for macroscopically examined systems
that are far from thermodynamic equilibrium.
There have been suggestions of additional laws, but none of them achieve the generality of the four
accepted laws, and they are not mentioned in standard textbooks.[1][2][3][4][5][8][9]
The laws of thermodynamics are important fundamental laws in physics and they are applicable in other
natural sciences.
Table of Contents
1 Zeroth law
2 First law
3 Second law
4 Third law
5 History
6 See also
7 References
8 Further reading
Zeroth law](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-79-320.jpg)


![spontaneously be conducted or radiated only from a higher-temperature region to a lower-temperature
region, but not the other way around.
The second law refers to a wide variety of processes, reversible and irreversible. Its main import is to tell
about irreversibility.
The prime example of irreversibility is in the transfer of heat by conduction or radiation. It was known long
before the discovery of the notion of entropy that when two bodies of different temperatures are
connected with each other by purely thermal connection, conductive or radiative, then heat always flows
from the hotter body to the colder one. This fact is part of the basic idea of heat, and is related also to the
so-called zeroth law, though the textbooks' statements of the zeroth law are usually reticent about that,
because they have been influenced by Carathéodory's basing his axiomatics on the law of conservation
of energy and trying to make heat seem a theoretically derivative concept instead of an axiomatically
accepted one. Šilahvý (1997) notes that Carathéodory's approach does not work for the description of
irreversible processes that involve both heat conduction and conversion of kinetic energy into internal
energy by viscosity (which is another prime example of irreversibility), because "the mechanical power
and the rate of heating are not expressible as differential forms in the 'external parameters'". [10]
The second law tells also about kinds of irreversibility other than heat transfer, and the notion of entropy
is needed to provide that wider scope of the law.
According to the second law of thermodynamics, in a reversible heat transfer, an element of heat
transferred, δQ, is the product of the temperature (T), both of the system and of the sources or
destination of the heat, with the increment (dS) of the system's conjugate variable, its entropy (S)
View formula on Wikipedia[1]
The second law defines entropy, which may be viewed not only as a macroscopic variable of classical
thermodynamics, but may also be viewed as a measure of deficiency of physical information about the
microscopic details of the motion and configuration of the system, given only predictable experimental
reproducibility of bulk or macroscopic behavior as specified by macroscopic variables that allow the
distinction to be made between heat and work. More exactly, the law asserts that for two given
macroscopically specified states of a system, there is a quantity called the difference of entropy between
them. The entropy difference tells how much additional microscopic physical information is needed to
specify one of the macroscopically specified states, given the macroscopic specification of the other,
which is often a conveniently chosen reference state. It is often convenient to presuppose the reference
state and not to explicitly state it. A final condition of a natural process always contains microscopically
specifiable effects which are not fully and exactly predictable from the macroscopic specification of the
initial condition of the process. This is why entropy increases in natural processes. The entropy increase
tells how much extra microscopic information is needed to tell the final macroscopically specified state
from the initial macroscopically specified state.[11]
Third law
The third law of thermodynamics is sometimes stated as follows:
The entropy of a perfect crystal at absolute zero is exactly equal to zero.
At zero temperature the system must be in a state with the minimum thermal energy. This statement
holds true if the perfect crystal has only one state with minimum energy. Entropy is related to the number
of possible microstates according to S = kBln(Ω), where S is the entropy of the system, kB Boltzmann's
constant, and Ω the number of microstates (e.g. possible configurations of atoms). At absolute zero there
is only 1 microstate possible (Ω=1) and ln(1) = 0.](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-82-320.jpg)

![V ∂p
1
Thermal expansion α=
V
· Free entropy
Internal energy U(S,V)
Enthalpy H(S,p) = U + pV
Helmholtz free energy A(T,V) = U − TS
Gibbs free energy G(T,p) = H − TS
The four laws of thermodynamics define fundamental physical quantities (temperature, energy, and
entropy) that characterize thermodynamic systems. The laws describe how these quantities behave
under various circumstances, and forbid certain phenomena (such as perpetual motion).
The four laws of thermodynamics are:1][2][3][4][5][6]
Zeroth law of thermodynamics: If two systems are in thermal equilibrium with a third system, they must
be in thermal equilibrium with each other. This law helps define the notion of temperature.
First law of thermodynamics: Heat and work are forms of energy transfer. Energy is invariably conserved
but the internal energy of a closed system changes as heat and work are transferred in or out of it.
Equivalently, perpetual motion machines of the first kind are impossible.
Second law of thermodynamics: The entropy of any isolated system not in thermal equilibrium almost
always increases. Isolated systems spontaneously evolve towards thermal equilibrium—the state of
maximum entropy of the system—in a process known as "thermalization". Equivalently, perpetual
motion machines of the second kind are impossible.
Third law of thermodynamics: The entropy of a system approaches a constant value as the temperature
approaches zero. The entropy of a system at absolute zero is typically zero, and in all cases is
determined only by the number of different ground states it has. Specifically, the entropy of a pure
crystalline substance at absolute zero temperature is zero.
Classical thermodynamics describes the exchange of work and heat between systems. It has a special
interest in systems that are individually in states of thermodynamic equilibrium. Thermodynamic](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-84-320.jpg)
![equilibrium is a condition of systems which are adequately described by only macroscopic variables.
Every physical system, however, when microscopically examined, shows apparently random microscopic
statistical fluctuations in its thermodynamic variables of state (entropy, temperature, pressure, etc.).
These microscopic fluctuations are negligible for systems which are nearly in thermodynamic
equilibrium and which are only macroscopically examined. They become important, however, for
systems which are nearly in thermodynamic equilibrium when they are microscopically examined, and,
exceptionally, for macroscopically examined systems that are in critical states,[7] and for
macroscopically examined systems that are far from thermodynamic equilibrium.
There have been suggestions of additional laws, but none of them achieve the generality of the four
accepted laws, and they are not mentioned in standard textbooks.[1][2][3][4][5][8][9]
The laws of thermodynamics are important fundamental laws in physics and they are applicable in other
natural sciences.
Zeroth law
The zeroth law of thermodynamics may be stated as follows:
If system A and system B are individually in thermal equilibrium with system C, then system A is in
thermal equilibrium with system B
The zeroth law implies that thermal equilibrium, viewed as a binary relation, is a Euclidean relation. If
we assume that the binary relationship is also reflexive, then it follows that thermal equilibrium is an
equivalence relation. Equivalence relations are also transitive and symmetric. The symmetric
relationship allows one to speak of two systems being "in thermal equilibrium with each other", which
gives rise to a simpler statement of the zeroth law:
If two systems are in thermal equilibrium with a third, they are in thermal equilibrium with each other
However, this statement requires the implicit assumption of both symmetry and reflexivity, rather than
reflexivity alone.
The law is also a statement about measurability. To this effect the law allows the establishment of an
empirical parameter, the temperature, as a property of a system such that systems in equilibrium with
each other have the same temperature. The notion of transitivity permits a system, for example a gas
thermometer, to be used as a device to measure the temperature of another system.
Although the concept of thermodynamic equilibrium is fundamental to thermodynamics and was clearly
stated in the nineteenth century, the desire to label its statement explicitly as a law was not widely felt
until Fowler and Planck stated it in the 1930s, long after the first, second, and third law were already
widely understood and recognized. Hence it was numbered the zeroth law. The importance of the law as](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-85-320.jpg)

![The second law of thermodynamics asserts the existence of a quantity called the entropy of a system
and further states that
When two isolated systems in separate but nearby regions of space, each in thermodynamic equilibrium
in itself (but not necessarily in equilibrium with each other at first) are at some time allowed to interact,
breaking the isolation that separates the two systems, allowing them to exchange matter or energy,
they will eventually reach a mutual thermodynamic equilibrium. The sum of the entropies of the initial,
isolated systems is less than or equal to the entropy of the final combination of exchanging systems. In
the process of reaching a new thermodynamic equilibrium, total entropy has increased, or at least has
not decreased.
It follows that the entropy of an isolated macroscopic system never decreases. The second law states
that spontaneous natural processes increase entropy overall, or in another formulation that heat can
spontaneously be conducted or radiated only from a higher-temperature region to a lower-temperature
region, but not the other way around.
The second law refers to a wide variety of processes, reversible and irreversible. Its main import is to tell
about irreversibility.
The prime example of irreversibility is in the transfer of heat by conduction or radiation. It was known
long before the discovery of the notion of entropy that when two bodies of different temperatures are
connected with each other by purely thermal connection, conductive or radiative, then heat always
flows from the hotter body to the colder one. This fact is part of the basic idea of heat, and is related
also to the so-called zeroth law, though the textbooks' statements of the zeroth law are usually reticent
about that, because they have been influenced by Carathéodory's basing his axiomatics on the law of
conservation of energy and trying to make heat seem a theoretically derivative concept instead of an
axiomatically accepted one. Šilahvý (1997) notes that Carathéodory's approach does not work for the
description of irreversible processes that involve both heat conduction and conversion of kinetic energy
into internal energy by viscosity (which is another prime example of irreversibility), because "the
mechanical power and the rate of heating are not expressible as differential forms in the 'external
parameters'".[10]
The second law tells also about kinds of irreversibility other than heat transfer, and the notion of
entropy is needed to provide that wider scope of the law.
According to the second law of thermodynamics, in a reversible heat transfer, an element of heat
transferred, δQ, is the product of the temperature (T), both of the system and of the sources or
destination of the heat, with the increment (dS) of the system's conjugate variable, its entropy (S)
View formula on Wikipedia[1]
The second law defines entropy, which may be viewed not only as a macroscopic variable of classical
thermodynamics, but may also be viewed as a measure of deficiency of physical information about the
microscopic details of the motion and configuration of the system, given only predictable experimental](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-87-320.jpg)
![reproducibility of bulk or macroscopic behavior as specified by macroscopic variables that allow the
distinction to be made between heat and work. More exactly, the law asserts that for two given
macroscopically specified states of a system, there is a quantity called the difference of entropy
between them. The entropy difference tells how much additional microscopic physical information is
needed to specify one of the macroscopically specified states, given the macroscopic specification of the
other, which is often a conveniently chosen reference state. It is often convenient to presuppose the
reference state and not to explicitly state it. A final condition of a natural process always contains
microscopically specifiable effects which are not fully and exactly predictable from the macroscopic
specification of the initial condition of the process. This is why entropy increases in natural processes.
The entropy increase tells how much extra microscopic information is needed to tell the final
macroscopically specified state from the initial macroscopically specified state.[11]
Third law
The third law of thermodynamics is sometimes stated as follows:
The entropy of a perfect crystal at absolute zero is exactly equal to zero.
At zero temperature the system must be in a state with the minimum thermal energy. This statement
holds true if the perfect crystal has only one state with minimum energy. Entropy is related to the
number of possible microstates according to S = kBln(Ω), where S is the entropy of the system, kB
Boltzmann's constant, and Ω the number of microstates (e.g. possible configurations of atoms). At
absolute zero there is only 1 microstate possible (Ω=1) and ln(1) = 0.
A more general form of the third law that applies to systems such as glasses that may have more than
one minimum energy state:
The entropy of a system approaches a constant value as the temperature approaches zero.
The constant value (not necessarily zero) is called the residual entropy of the system.
History
See also: Philosophy of thermal and statistical physics
Count Rumford (born Benjamin Thompson) showed, about 1797, that mechanical action can generate
indefinitely large amounts of heat, so challenging the caloric theory. The historically first established
thermodynamic principle which eventually became the second law of thermodynamics was formulated
by Sadi Carnot during 1824. By 1860, as formalized in the works of those such as Rudolf Clausius and
William Thomson, two established principles of thermodynamics had evolved, the first principle and the
second principle, later restated as thermodynamic laws. By 1873, for example, thermodynamicist Josiah
Willard Gibbs, in his memoir Graphical Methods in the Thermodynamics of Fluids, clearly stated the first
two absolute laws of thermodynamics. Some textbooks throughout the 20th century have numbered
the laws differently. In some fields removed from chemistry, the second law was considered to deal with
the efficiency of heat engines only, whereas what was called the third law dealt with entropy increases.](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-88-320.jpg)






















![528
Selected References
These references are in PubMed. This may not be the complete list of references from this article.
Bird HL, Pugh CT. A Paper Chromatographic Separation of Gibberellic Acid and Gibberellin A. Plant
Physiol. 1958 Jan;33(1):45–46. [PMC free article] [PubMed]
DARKEN MA, JENSEN AL, SHU P. Production of gibberellic acid by fermentation. Appl Microbiol. 1959
Sep;7:301–303. [PMC free article] [PubMed]
STODOLA FH, RAPER KB, FENNELL DI, CONWAY HF, SOHNS VE, LANGFORD CT, JACKSON RW. The
microbiological production of gibberellins A and X. Arch Biochem Biophys. 1955 Jan;54(1):240–245.
[PubMed]
Articles from Applied Microbiology are provided here courtesy of American Society for Microbiology
(ASM)
Formats:
Summary
|
Page Browse
|
PDF (923K)
Related citations in PubMed
Gibberellic acid production by Fusarium moniliforme on lupin seed extract. [Acta Microbiol Pol. 1994]
Gibberellic acid production by Fusarium moniliforme on lupin seed extract.
Gulewicz K, Rataj-Guranowska M, Lukaszewska N, Michalski Z. Acta Microbiol Pol. 1994; 43(1):73-7.
Survey of some Fusarium moniliforme strains from different host plants for compounds possessing
gibberellin-like activity. [Zentralbl Bakteriol Parasitenkd Infektionsk...]](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-113-320.jpg)
![Survey of some Fusarium moniliforme strains from different host plants for compounds possessing
gibberellin-like activity.
El-Bahrawi S. Zentralbl Bakteriol Parasitenkd Infektionskr Hyg. 1977; 132(2):178-83.
Biosynthesis of gibberellic acid from milk permeate in repeated batch operation by a mutant Fusarium
moniliforme cells immobilized on loofa sponge. [Bioresour Technol. 2009]
Biosynthesis of gibberellic acid from milk permeate in repeated batch operation by a mutant Fusarium
moniliforme cells immobilized on loofa sponge.
Meleigy SA, Khalaf MA. Bioresour Technol. 2009 Jan; 100(1):374-9. Epub 2008 Aug 5.
Morphological mutants of Gibberella fujikuroi for enhanced production of gibberellic acid. [J Appl
Microbiol. 2006]
Morphological mutants of Gibberella fujikuroi for enhanced production of gibberellic acid.
Lale G, Jogdand VV, Gadre RV. J Appl Microbiol. 2006; 100(1):65-72.
EFFECTS OF CORN STEEP LIQUOR AND THIAMINE ON L-GLUTAMIC ACID FERMENTATION OF
HYDROCARBONS.IV. UTILIZATION OF HYDROCARBONS BY MICROORGANISMS. [Appl Microbiol. 1965]
EFFECTS OF CORN STEEP LIQUOR AND THIAMINE ON L-GLUTAMIC ACID FERMENTATION OF
HYDROCARBONS.IV. UTILIZATION OF HYDROCARBONS BY MICROORGANISMS.
TAKAHASHI J, KOBAYASHI K, IMADA Y, YAMADA K. Appl Microbiol. 1965 Jan; 13:1-4.
See reviews... See all...
Links
PubMed
PubMed
PubMed citation for this article
Substance
Substance
PubChem Substance links
Taxonomy
Taxonomy
Related taxonomy entry](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-114-320.jpg)














![IUPAC name[show]
Identifiers
CAS number 77-06-5
PubChem 522636
ChemSpider 7995349
UNII BU0A7MWB6L
EC number 201-001-0
KEGG C01699
ChEBI CHEBI:28833
ChEMBL CHEMBL566653
Jmol-3D images Image 1
SMILES
[show]
InChI
[show]
Properties
Molecular formula C19H22O6](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-135-320.jpg)
![Molar mass 346.37 g/mol
Melting point 233 - 235 °C (decomposition)
Solubility in water 5 g/l (20 °C)
Hazards
EU classification Irritant (Xi)
R-phrases R36
S-phrases R26, S36
(verify) (what is: / ?)
Except where noted otherwise, data are given for materials
in their standard state (at 25 °C, 100 kPa)
Infobox references
Gibberellic acid (also called Gibberellin A3, GA, and GA3) is a hormone found in plants. Its
chemical formula is C19H22O6. When purified, it is a white to pale-yellow solid.
Gibberellic acid is a simple gibberellin, a pentacyclic diterpene acid promoting growth and
elongation of cells. It affects decomposition of plants and helps plants grow if used in small
amounts, but eventually plants develop tolerance to it[citation needed]. GA stimulates the cells of
germinating seeds to produce mRNA molecules that code for hydrolytic enzymes. Gibberellic
acid is a very potent hormone whose natural occurrence in plants controls their development.
Since GA regulates growth, applications of very low concentrations can have a profound effect
while too much will have the opposite effect.[1] It is usually used in concentrations between 0.01
and 10 mg/L.
GA was first identified in Japan in 1935, as a metabolic byproduct of the plant pathogen
Gibberella fujikuroi (thus the name), which afflicts rice plants; fujikuroi-infected plants develop
bakanae ("foolish seedling"), which causes them to grow so much taller than normal that they
die from no longer being sturdy enough to support their own weight.
Gibberellins have a number of effects on plant development. They can stimulate rapid stem and
root growth, induce mitotic division in the leaves of some plants, and increase seed germination
rate.](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-136-320.jpg)
![Gibberellic acid is sometimes used in laboratory and greenhouse settings to trigger germination
in seeds that would otherwise remain dormant.[1] It is also widely used in the grape-growing
industry as a hormone to induce the production of larger bundles and bigger grapes, especially
Thompson seedless grapes. In the Okanagan and Creston valleys, it is also used as a growth
replicator in the cherry industry.
[edit] See also
Gibberellin
Plant hormone
6-Benzylaminopurine
[edit] References
1. ^ a b Riley, John M.. "Gibberellic Acid for Fruit Set and Seed Germination".
http://www.crfg.org/tidbits/gibberellic.html. Retrieved 26 Oct 2012.
[edit] External links
No longer maintained. Safety MSDS data
Synonyms at webbook.nist.gov
www.crfg.org
Retrieved from "http://en.wikipedia.org/w/index.php?title=Gibberellic_acid&oldid=535424207"
View page ratings
Rate this page
What's this?
Trustworthy
Objective
Complete
Well-written
I am highly knowledgeable about this topic (optional)](https://image.slidesharecdn.com/krishna-130129033122-phpapp01/85/Krishna-137-320.jpg)