Downloaded 64 times




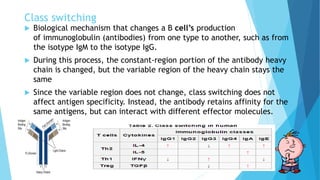
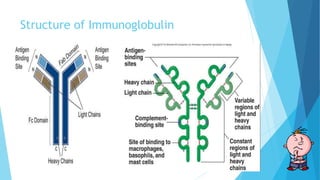
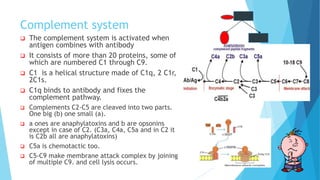



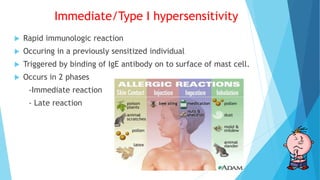
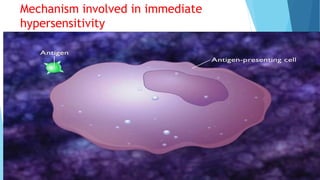
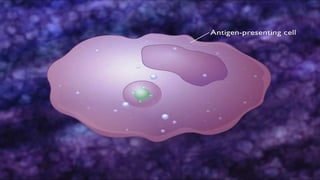
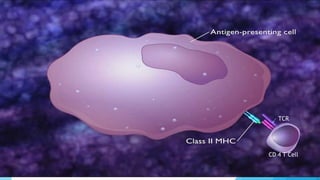
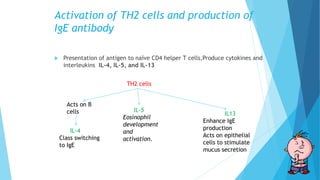
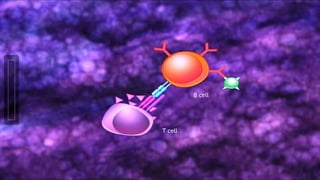
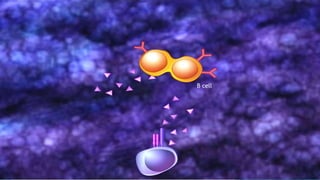
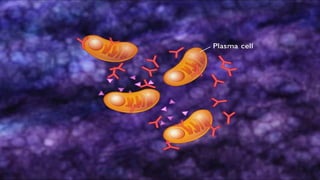
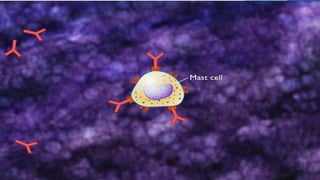
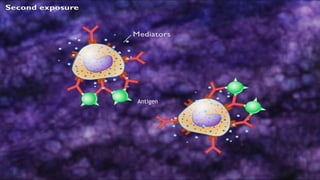
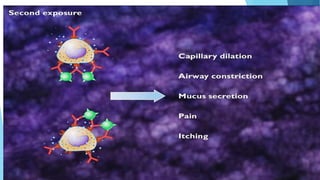
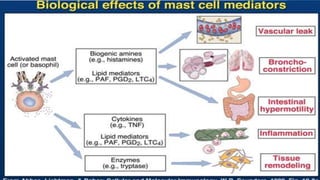
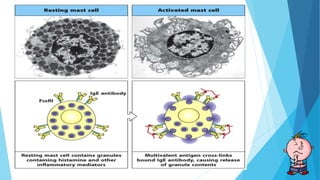



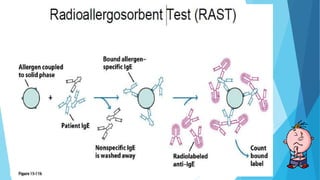


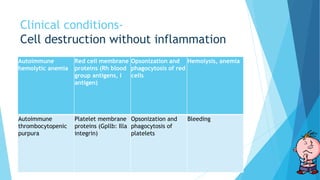
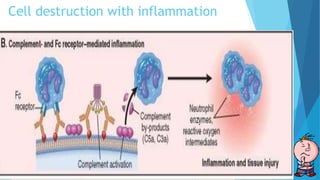
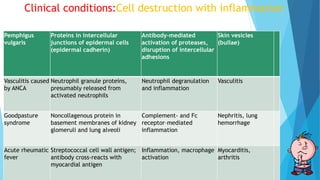
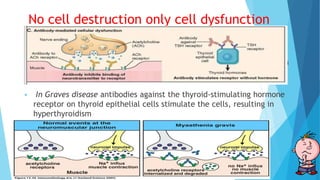



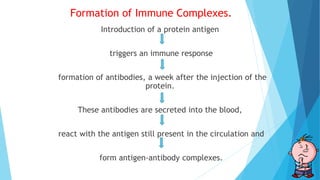

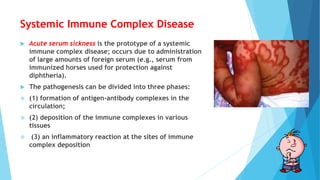
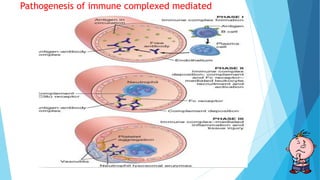
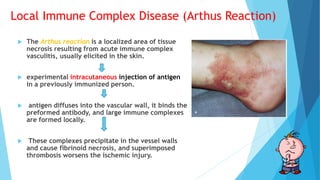
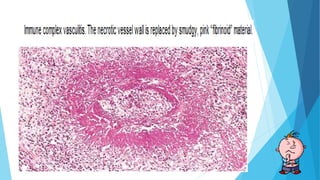

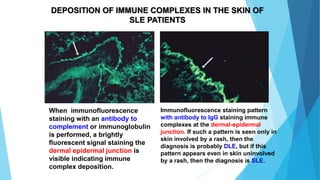

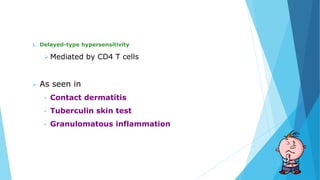
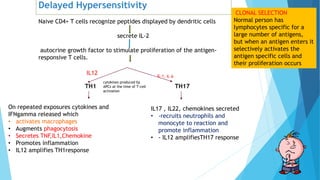
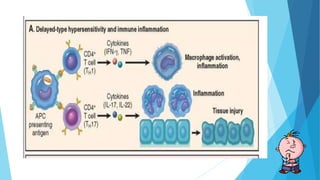
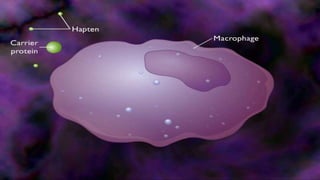
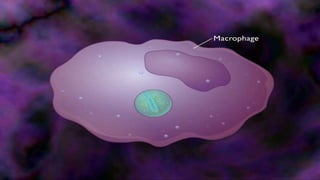
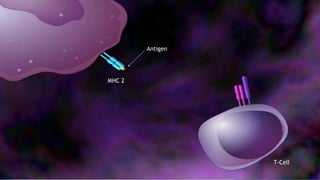
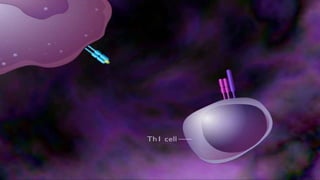
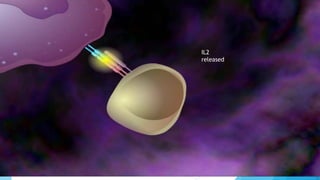
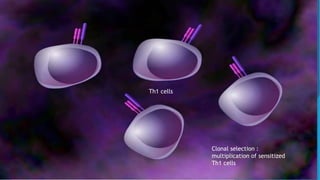

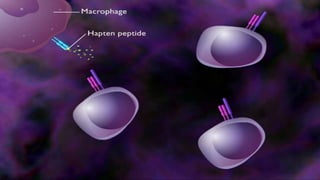
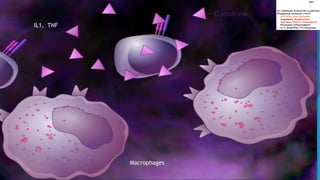
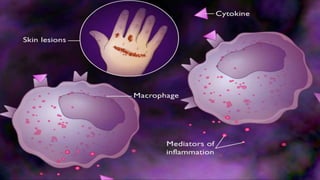
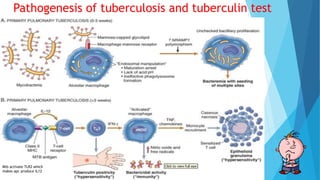
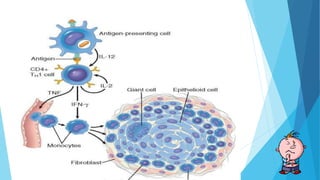
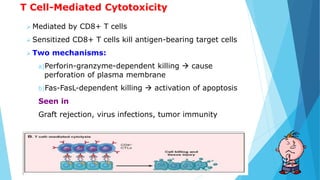
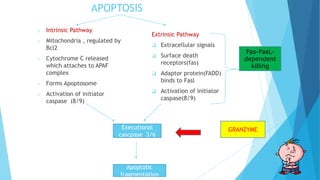
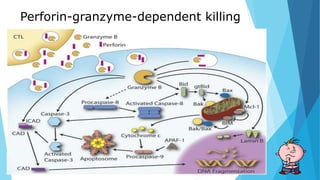
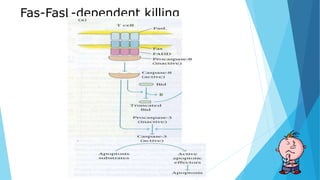
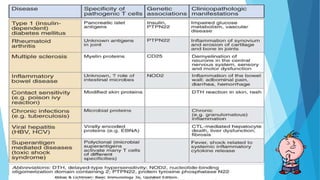




Hypersensitivity reactions are harmful immune responses that occur upon reexposure to an antigen in sensitized individuals. There are four main types of hypersensitivity: Type I is immediate and involves IgE antibodies and mast cells/basophils; Type II involves antibody-antigen interactions on cell surfaces; Type III involves immune complex deposition; Type IV is cell-mediated and delayed. The document defines key terms and outlines the mechanisms, mediators, clinical manifestations of each hypersensitivity type.



































































