




























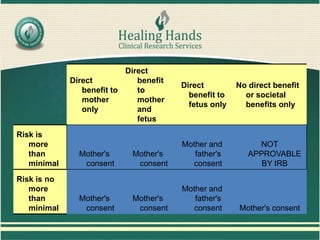















Vulnerable populations include groups who may have impaired ability to provide fully informed consent to participate in clinical trials. These include children, pregnant women, prisoners, students/employees, and those with cognitive impairments. Additional protections are required when including such groups in research. For children, assent from the child and permission from parents/guardians is needed. Research involving pregnant women or fetuses generally requires the mother's consent and may require the father's consent depending on the study's risks and benefits. Prisoners can only be in research related to their incarceration or behavior with minimal risk. Cognitively impaired individuals may require consent from a legally authorized representative. Researchers must consider risks unique to vulnerable populations and implement safeguards to



































































