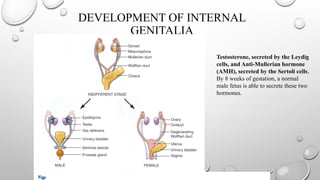
DEVELOPMENT OF INTERNAL
GENITALIA
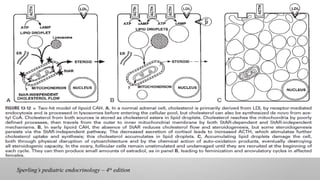
Testosterone,secreted by the Leydig
cells, and Anti-Mullerian hormone
(AMH), secreted by the Sertoli cells.
By 8 weeks of gestation, a normal
male fetus is able to secrete these two
hormones.
5.
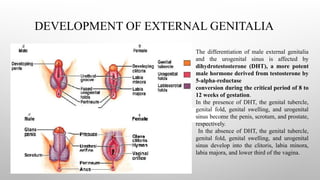
DEVELOPMENT OF EXTERNALGENITALIA
The differentiation of male external genitalia
and the urogenital sinus is affected by
dihydrotestosterone (DHT), a more potent
male hormone derived from testosterone by
5-alpha-reductase
conversion during the critical period of 8 to
12 weeks of gestation.
In the presence of DHT, the genital tubercle,
genital fold, genital swelling, and urogenital
sinus become the penis, scrotum, and prostate,
respectively.
In the absence of DHT, the genital tubercle,
genital fold, genital swelling, and urogenital
sinus develop into the clitoris, labia minora,
labia majora, and lower third of the vagina.
6.
CONGENITAL ADRENAL HYPERPLASIA
•Family of autosomal recessive disorders affecting adrenal steroidogenesis
• 21-hydroxylase deficiency
• 11β-hydroxylase deficiency
• 17α-hydroxylase deficiency
• 3β-hydroxysteroid dehydrogenase deficiency
• Lipoid/star CAH
• U.S. Occurrences – 1:15,500 Caucasian births, 1:42,000 African American births
• Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder with an incidence ranging from 1:10,000 to 1:20,000
births1
.
• The screen positive rate of CAH among a cohort of 104,066 babies screened at birth in India was 1 in 5762 as per a recent report2
1. Therrell B. Newborn screening for congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. 2001;30:15-30
2. ICMR Task Force on Inherited Metabolic Disorders. Newborn screening for congenital hypothyroidism and congenital adrenal hyperplasia. Indian J
Pediatr. 2018;85:935-40
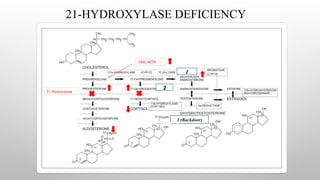
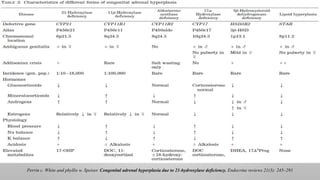
21-HYDROXYLASE DEFICIENCY
• Accountsfor 95% of all cases. (1 in 14,000, heterozygous carrier
rate of 1 in 60.) CYP21A2 gene on Chr. 6
• Classic type
• 1) Salt Wasting Form
• 2) Simple Virilizing Form
• ◦ Non-classic type
• More than 90% of these mutations result from intergenic
recombinations between CYP21 and the closely linked CYP21P
pseudogene.
• Approximately 20% are gene deletions due to unequal crossing
over during meiosis,
• Whereas the remainder are gene conversions—transfers to CYP21
of deleterious Mutations normally present in CYP21P
Classic Phenotype Non-classic
Phenotype
Severe form Mild/late-onset form
1:15,000 live births 1:1,000 live births
Salt-losing (67%) or Simple
virilizing (33%)
9.
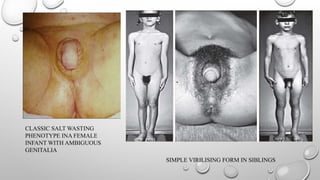
Classic 21-Hydroxylase DeficiencyExam Findings
FEMALES MALES
• Enlarged clitoris
• Partly-fused, rugose labia majora
• Common urogenital sinus in place of
urethra and vagina
• Salt-losing present soon after birth,
given ambiguous genitalia
• Normal internal female organs.
• Most common cause of 46XX DSD
• Subtle hyperpigmentation
• Possible penile enlargement
• Salt-losing presents DOL 7-14
with emesis, weight loss, lethargy,
dehydration, shock, hyponatremia,
hyperkalemia
• Non-salt-losing present with early
virilization at 2-4yrs
• Normal male internal organs, but
can have small testes if untreated.
SALT-WASTING 21-HYDROXYLASE
DEFICIENCY
• 67%patients with classic 21-hydroxylase deficiency
• Secondary to aldosterone deficiency
• Associated lab abnormalities: hyponatremia, hyperkalemia
• Early signs: frequent feedings
• Present with salt-wasting and acute adrenal crisis within weeks after
birth. (usually the second week)
• About 75% of Classic CAH cases suffer aldosterone deficiency with
Salt wasting, vomiting, diarrhea, failure to thrive, and potentially fatal
Hypovolemia and shock
SIMPLE VIRILIZING FORM
•No salt wasting
• Unless detected by newborn screening, males with this disorder may escape diagnosis until
age 3 to 7 years, when they come to medical attention because of early development of pubic,
axillary, and facial hair; acne; and phallic growth
• Untreated or poorly treated children with CAH may fail to undergo normal puberty, and boys
may have small testes and azoospermia because of the feedback of the adrenally produced
testosterone on pituitary gonadotropins
• High concentrations of ACTH in poorly treated males may stimulate the enlargement of adrenal
rests in the testes. These enlarged testes usually appear nodular by ultrasound, unlike the
homogeneously enlarged testes in central precocious puberty.
Sperling’s Pediatric Endocrinology – 4th
Edition
15.
NON-CLASSIC 21-HYDROXYLASE DEFICIENCY
EXAMFINDINGS
• Present in late childhood or early adulthood
• Hyperandrogenism manifestations
• Premature adrenarche
• Advanced bone age, short adult stature
• Female hirsutism (60%) or male-pattern baldness
• Oligomenorrhea/amenorrhea (54%)
• Polycystic ovaries
• Insulin resistance
• Acne (33%)
• 5-10% children with premature adrenarche have an underlying diagnosis of non-classic
CAH
PRENATAL DIAGNOSIS/TREATMENT
• Thecriteria for considering prenatal steroids are
• history of previously affected sibling or first degree relative with known mutations,
• period of gestation less than 9 weeks,
• aim to continue pregnancy till term with good drug compliance
• Dexamethasone 5-6wks gestation : The usual protocol employs dexamethasone doses of 20 mg/kg of maternal body
weight (with a maximum dose of 1.5 mg/day). 70% of treated female fetuses are born with normal or only minimally
virilized genitalia.
• Chromosome analysis : Chorionic villous sampling (CVS) at 9-12wks; Amniocentesis at 14-18wks
• If male, discontinue steroids. If female, pursue additional molecular testing. If affected, continue dexamethasone
to term. If both parents have classic CAH, the risk of having a daughter with CAH is 1:8.
• The adverse fetal outcomes reported are spontaneous abortion, fetal demise, intrauterine growth retardation, liver
steatosis and congenital malformations. Mild cognitive and behavioral abnormalities have been reported in children
who received prenatal steroids.
• Fetal sex and CYP21A2 genotype can be determined as early as week 6 of gestation with the use of novel molecular
diagnostic methods that analyze cell-free fetal DNA from maternal blood using real-time polymerase chain reaction.
In this way, the number of unnecessarily treated cases can be substantially reduced.
18.
INITIAL LABORATORY WORK-UP
•Glucose
• Serum electrolytes
• ABG
• Cortisol level
• ACTH
• 17-hydroxyprogesterone level
• Pelvic/scrotal ultrasound
• Karyotype
• Serum androgens & Urinary 17 Ketosteroids (24 hr urine
sample mandatory with concomitant urine creatinine)
• Plasma Renin activity to aldosterone ratio
• Newborn screens
• High 17-hydroxyprogesterone (17-OHP)
level >242 nmol/L on day of life 3.
• A complete adrenocortical profile is
recommended to differentiate 21-OH
deficiency from other enzyme defects and to
diagnose borderline cases.
• Serum 17-OHP, cortisol, 11-deoxycortisol, 17-
OH pregnenolone, dehydroepiandrostenedione
and androstenedione should be measured to
differentiate.
Reserve adrenal imaging for individuals with classic congenital adrenal hyperplasia who have clinical evidence of an adrenal mass, poor
disease control, a lapse in treatment of several years, or lack of response to intensified therapy.
19.
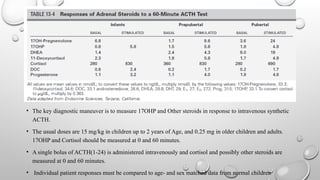
• The keydiagnostic maneuver is to measure 17OHP and Other steroids in response to intravenous synthetic
ACTH.
• The usual doses are 15 mg/kg in children up to 2 years of Age, and 0.25 mg in older children and adults.
17OHP and Cortisol should be measured at 0 and 60 minutes.
• A single bolus of ACTH(1-24) is administered intravenously and cortisol and possibly other steroids are
measured at 0 and 60 minutes.
• Individual patient responses must be compared to age- and sex matched data from normal children
20.
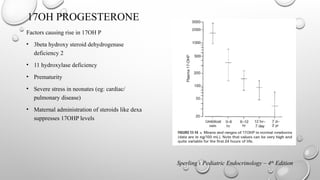
17OH PROGESTERONE
Factors causingrise in 17OH P
• 3beta hydroxy steroid dehydrogenase
deficiency 2
• 11 hydroxylase deficiency
• Prematurity
• Severe stress in neonates (eg: cardiac/
pulmonary disease)
• Maternal administration of steroids like dexa
suppresses 17OHP levels
Sperling’s Pediatric Endocrinology – 4th
Edition
21.
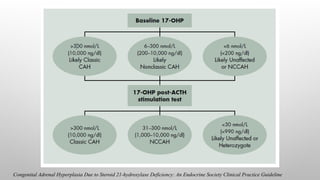
17OHP LEVELS
The differentforms of CAH due to
21OHD may be determined based on
baseline and ACTH stimulated values
of 17-OHP, the immediate precursor
to the 21-hydroxylase enzyme,
although overlaps may exist.
Sperling’s Pediatric Endocrinology – 4th
Edition
22.
Congenital Adrenal HyperplasiaDue to Steroid 21-hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline
23.
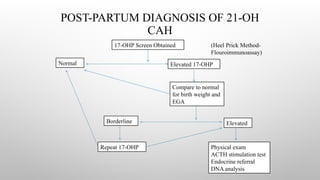
POST-PARTUM DIAGNOSIS OF21-OH
CAH
17-OHP Screen Obtained
Normal Elevated 17-OHP
Compare to normal
for birth weight and
EGA
Borderline Elevated
Repeat 17-OHP Physical exam
ACTH stimulation test
Endocrine referral
DNA analysis
(Heel Prick Method-
Flouroimmunoassay)
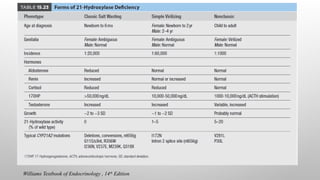
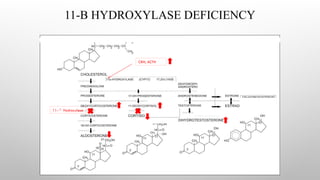
11-B-HYDROXYLASE DEFICIENCY
• Accountsfor 5-10% of cases of CAH.
• Gene is located on the long arm of chromosome 8.
• It is characterized by low plasma renin activity & elevation of serum 11-
deoxycortisol and 11-deoxycorticosterone.
• Because of the strong mineralocorticoid activity of deoxycorticosterone, the
condition is characterized by salt retention, hypertension & hypokalemic alkalosis.
• The elevated plasma androgens may cause virilization of the female fetus.
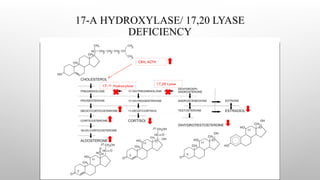
17-A-HYDROXYLASE DEFICIENCY
• Geneticdefect is on chromosome 10; 17A Ohlase deficiency is common in Brazil.
• These Patients may have mild symptoms of glucocorticoid deficiency, But this is not life threatening, as the lack of P450c17
results in the overproduction of corticosterone, which also has glucocorticoid activity.
• Presents with similar features of those of 11-hydroxylase deficiency except that androgens are low, so no virilization in girls &
genitalia is ambiguous in boys.
• Affected males are incompletely virilized and present as phenotypic females (but gonads are usually palpable in the Inguinal
region or the labia : Male pseudohermaphroditism) or with sexual ambiguity.
• Affected females usually present with failure of sexual development at the expected time of puberty due to absence of
steroidogenesis in gonads.
• The diagnosis is made by finding low or absent 17-hydroxylated C-21 and C-19 plasma steroids, which respond
poorly to stimulation with ACTH
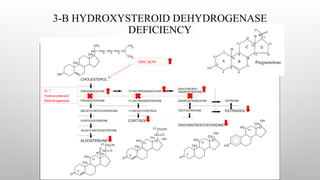
3-B-HYDROXYSTEROID DEHYDROGENASE TYPE2
DEFICIENCY
• This is a very rare disorder that results in accumulation of DHEA, which is converted to testosterone in
peripheral tissues.
• Because cortisol and aldosterone are not synthesized in patients with the classic form of the disease, infants
are prone to salt-wasting crises.
• Because androstenedione and testosterone are not synthesized, boys are incompletely virilized. Varying
degrees of hypospadias may occur, with or without bifid scrotum or cryptorchidism.
• Because DHEA levels are elevated and this hormone is a weak androgen, girls are mildly virilized, with
slight to moderate clitoral enlargement
• Thus, the principal Diagnostic test in 3βHSD 2 deficiency is intravenous administration Of ACTH
with measurement of the ratio between three Δ5 compounds and the corresponding Δ4 compounds.
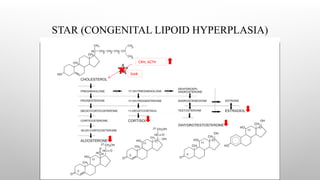
CONGENITAL LIPOID HYPERPLASIA
Patients have normal p450scc, with normal ferrodoxin reductase & ferrodoxin. Placental steroidogenesis intact as it does
not require STAR protein.
Patients with lipoid adrenal hyperplasia are usually unable to synthesize any adrenal steroids. Thus, affected infants are
likely to be confused with those with adrenal hypoplasia congenita.
Salt-losing manifestations are typical manifesting in first few weeks and many infants die in early infancy.
Genetic males are unable to synthesize androgens and thus are phenotypically female but with gonads palpable in the
labia majora or inguinal areas.
Genetic Females appear normal at birth and may undergo feminization at puberty with menstrual bleeding. They too,
progress to hypergonadotropic Hypogonadism when accumulated cholesterol kills granulosa (I.E., Steroid synthesizing)
cells in the ovary.
33.
, mild virilization
infemales
Perrin c. White and phyllis w. Speiser. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocrine reviews 21(3): 245–291
34.
DIFFERENTIAL DIAGNOSIS
• Adrenalhypoplasia
• Bilateral adrenal hemorrhage
• Familial glucocorticoid deficiency
• Disorders of sex development
• Adrenal insufficiency
• PCOS
35.
MANAGEMENT
• The goalof therapy is to reduce excessive androgen secretion by replacing the deficient
hormones.
• Proper treatment with glucocorticoids prevents adrenal crisis and virilization, allowing normal
growth and development.
• Clinical management of classic CAH is a difficult balance between hyperandrogenism and
hypercortisolism.
• Undertreatment carries the risk of adrenal crisis and allows increased adrenal androgen
production, with accelerated bone age and loss of growth potential.
• Overtreatment may suppress growth, increase blood pressure, and cause iatrogenic Cushing’s
syndrome.
36.
MANAGEMENT OF ACUTEADRENAL CRISIS
Patients experiencing an adrenal crisis virtually always have significant volume contraction,
and initial management should consist of fluid resuscitation with a 20-ml/kg Bolus of 0.9%
normal saline. Repeated boluses may be needed.
Replacement of fluid losses should be continued With isotonic crystalloid solutions containing
dextrose (Typically 5% dextrose with normal saline) at a rate of 1.5 to 2 times maintenance.
Stress doses of hydrocortisone (100 mg/m2 per day) are vital in the management and should
be given as early as possible, concomitant with intravenous (IV) fluid treatment.
37.
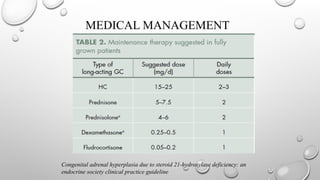
MEDICAL MANAGEMENT
Patients whohave classic CAH cannot mount a sufficient Cortisol response to stress and
require pharmacologic doses of hydrocortisone in situations such as febrile Illness, trauma, and
surgery under general anesthesia.
During times of mild-to-moderate stress, such as fever, Dosing guidelines suggest doubling or
tripling the usual Maintenance dose of oral hydrocortisone.
PRA can be used to monitor the effectiveness of mineralocorticoid and sodium replacement.
Hypertension, tachycardia, and suppressed PRA production are clinical signs of overtreatment.
Treatment efficacy is assessed by monitoring ACTH, DHEA, and androstenedione. Each visit
should be accompanied by measurement of blood pressure, plasma renin activity and serum δ4-
androstenedione, DHEA, DHEA sulfate, and testosterone.
Plasma 17ohp is generally measured but may be difficult to interpret because of its variation as a
function of the timing of glucocorticoid doses, its diurnal variation and its hyperresponsiveness to
stress (e.x., Clinic visits)
38.
MEDICAL MANAGEMENT
Patients shouldbe monitored carefully for signs of iatrogenic Cushing syndrome, including
Rapid weight gain, poor growth velocity, hypertension, Pigmented striae, and osteopenia.
• Measurements of growth should be made at 3- to 4-month intervals in children, along with an
annual assessment of bone age. Children also should have an annual bone age radiograph and
careful monitoring of linear growth. Height was most affected in SW-CAH than SV-CAH and in
children less than two years than in older age8
.
• Advanced bone age and increased Growth velocity should alert pediatricians to
undertreatment.
8. Meena H, Jana M, Singh V, Kabra M, Jain V. Growth pattern and clinical profile of Indian children with classical 21-hydroxylase deficiency
congenital adrenal hyperplasia on treatment. Indian J Pediatr. 2019;Jan 30
39.
Dabas et al,Management of infants with congenital adrenal hyperplasia. Indian pediatr 2020;57: 159-164
Follow up of CAH patients
Hormones Should be measured, preferably at 8 AM, just before the Next dose is due. The hormone evaluation can be
performed in urine, blood, saliva or dried blood filter paper. In patients ≤18 months with congenital adrenal hyperplasia,
recommended close monitoring in the first 3 months of life and every 3 months thereafter. After 18 months, we
recommend evaluation every 4 months.
MEDICAL MANAGEMENT
Mostauthorities favor the use of oral hydrocortisone or cortisone acetate in three divided daily
doses in growing children
In older children and adolescents, where growth is complete, once-daily dexamethasone offers
a simpler dosing regimen and may be considered instead of Hydrocortisone for improved
compliance.
LINEAR GROWTH
• Childrenwith classic CAH have accelerated linear growth, but adult height is 1-2 standard
deviations below mean target height.
• The loss of height in CAH is partially due to the effect of sex steroids on epiphyseal closure and
partially due to glucocorticoid-induced resistance to the action of growth hormone; the most
crucial times when height is lost are during the first 2 years of life and during the pubertal growth
spurt
• Recommend use of lowest effective treatment dose to maintain hormone levels, vary with age.
• Non-classic CAH adult height consistent with mean parental height if appropriately treated.
44.
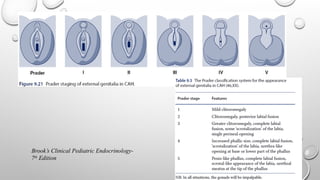
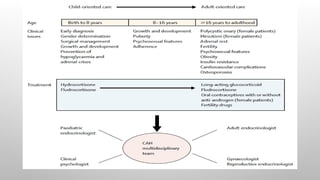
GENDER ASSIGNMENT
• Genderassignment may be difficult, and not always possible, immediately after birth.
• Gender assignment should be done after expert opinion and appropriate counseling and discussion with
the parents.
• In patients with 46 XX DSD due to 21-hydroxylase deficiency, gender identity is generally female and
fertility is possible. Hence, according to an international consensus guideline, female gender assignment is
advised10
.
10. Lee PA, houk CP, ahmed SF, hughes IA. Consensus statement on management of intersex disorders.
International consensus conference on intersex. Pediatrics. 2006;118:e488-500
Transition to adult care should begin several years prior to dismissal from pediatric endocrinology.
The use of joint clinics comprised of pediatric, reproductive, and adult endocrinologists and urologist during this transition
should be made available
45.
GENITALIA RECONSTRUCTIVE SURGERY
•Decision of genital surgery needs to involve multi-disciplinary approach.
• The goals of corrective surgery are
• Improving the appearance of external genitalia to resemble normal female
genitalia,
• Conserve sexual and reproductive functions,
• Achieve adequate urinary stream without incontinence.
• The decision for corrective surgery should never be taken in haste during early
newborn period
• Procedures:
• Clitoroplasty, vaginoplasty & labial surgery are the commonest
• Partial clitoral reduction in infancy with vaginoplasty reserved for late adolescence.
• In general, procedures have improved, but overall outcome is not optimal.
46.
REPRODUCTIVE FUNCTION INFEMALES
• Delayed spontaneous menarche compared to peers, atrophic breast
tissue
• Poorly controlled females
• Hirsutism
• Oligomenorrhea, amenorrhea
• Menorrhagia
• Absence of thelarche, precocious adrenarche
• Cystic acne
• Cystic ovaries
• Fertility and offspring concerns
• Some evidence of infertility: inadequate vaginal introitus, poor adrenal
suppression, high prevalence of polycystic ovary syndrome, and elevated
follicular phase progesterone concentrations causing failure of implantation.
47.
REPRODUCTIVE FUNCTION INMALES
• Small testicular size
• Gonadal function:
• In males, high levels of ACTH can also stimulate formation of testicular adrenal rest tumors (TARTS),
which impair testicular function and can cause oligospermia
• Beyond five years of age, affected males should be screened for development of any TARTS by serial
ultrasonography of testis to detect hypoechoic lesions. Usually these lesions are small, not clinically
discernible, and regress with better titration of steroid therapy.
48.
PSYCHOSEXUAL CONCERNS
• Femaleswith CAH (especially salt-wasters)
• Male-typical play: Girls exposed to high amounts of prenatal androgen are more likely to
prefer “boy toys” such as cars and guns instead of “girl toys” such as dolls and kitchen
equipment.
• Physical aggression
• Low-interest in infants/maternal nurturing
• Most raised as female express female gender identity, gender role, and heterosexual orientation
• Increased rate of homosexuality compared to peers(5-37%)
• Males with CAH
• No evidence of atypical gender behavior reported
49.
DEVELOPMENTAL CONCERNS
• Overall,IQ is similar among patients with CAH and their matched controls.
• Some data show that poorly controlled salt-wasting children with CAH are prone to learning
disabilities.
51.
REFERENCES
1. Therrell B.Newborn screening for congenital adrenal hyperplasia. Endocrinol metab clin north am.
2001;30:15-30
2. ICMR task force on inherited metabolic disorders. Newborn screening for congenital hypothyroidism and
congenital adrenal hyperplasia. Indian J pediatr. 2018;85:935-40
3. Brook’s Clinical Pediatric Endocrinology- 7th
edition
4. Sperling’s pediatric endocrinology – 4th
edition
5. Williams Textbook Of Endocrinology- 14th
Edition
6. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical
practice guideline
7. Perrin c. White and phyllis w. Speiser. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency.
Endocrine reviews 21(3): 245–291
8. Meena H, jana M, singh V, kabra M, jain V. Growth pattern and clinical profile of indian children with
classical 21-hydroxylase deficiency congenital adrenal hyperplasia on treatment. Indian J pediatr.
2019;jan 30
52.
9. Dabas etal, management of infants with congenital adrenal hyperplasia. Indian pediatr
2020;57: 159-164.
10. Lee PA, houk CP, ahmed SF, hughes IA. Consensus statement on management of intersex
disorders. International consensus conference on intersex. Pediatrics. 2006;118:e488-500
REFERENCES
#9 The fetal testes produce large amounts of testosterone in early to midgestation, which differentiates the pluripotent embryonic precursor structures into male external genitalia. In the male fetus with 21-hydroxylase deficiency, the additional testosterone produced in the adrenals has no phenotypic effect. By contrast, the fetal ovaries normally produce no sex steroids or other factors
needed for differentiation of the female external genitalia. The testosterone inappropriately produced by the adrenals of the CAH female fetus causes varying degrees of external genital virilization.
#14 Because the adrenal normally produces 100 to 1000 times as much cortisol as aldosterone, mild defects (amino acid replacement mutations) in p450c21 are less likely to affect mineralocorticoid secretion than cortisol secretion ( No salt wasting)
These signs may be mistaken for premature puberty, but the astute physician can readily differentiate such boys with sexual precocity from boys with true central precocious puberty, as the testes remain of prepubertal size in CAH, whereas gonadotropic stimulation in true precocious puberty results in pubertal-sized testes ( 4 mL).
#17 Female fetuses affected with CAH begin to become virilized at about 6 to 8 weeks’ gestation, the same time at which the testes of normal male fetuses produce testosterone, causing fusion of the labioscrotal folds, enlargement of the genital tubercle into a phallus, and the formation of the phallic urethra. The adrenals of female fetuses with CAH may produce concentrations of testosterone that approach those of a normal male, resulting in varying degrees of masculinization of the external genitalia.
The rationale is that dexamethasone, which is not metabolized by placental 11bHSD2, will cross the placenta, suppress fetal ACTH, and consequently suppress adrenal steroidogenesis. Treatment
of a fetus with CAH requires the use of fluorinated steroids that escape metabolism by these enzymes.
The potential benefits of prenatal treatment are reduction or elimination of virilization of external genitalia and brain, reducing the risk of gender confusion and the need for surgery
#18 Urinary 17 ketosteroids are more useful in evaluating the efficacy of treatment.
Plasma renin activity and its response to salt restriction scan be especially useful.
Most patients with simple virilizing 21-hydroxylase deficiency have high plasma renin activity that increases further on sodium restriction, confirming that these patients are partially mineralocorticoid deficient and can maintain a normal serum sodium level only by hyperstimulation of the zona glomerulosa.
#19 Both basal and stimulated levels of 17OHP are markedly elevated in patients with salt-losing and simple virilizing forms of 21-hydroxylase deficiency. Basal levels are usually greater than 2000 ng/dL and increase to more than 5000 to 10,000 ng/dL after ACTH. Patients with the milder late-onset or cryptic forms typically have normal to mildly elevated basal levels but have supranormal responses to ACTH stimulation (i.e., 1500 to . 10,000 ng/dL). The cortisol response to ACTH is absent or subnormal in patients with the classic forms of CAH and is normal in patients with late-onset and cryptic forms.
Basal plasma ACTH levels reflect the extent of 21-hydroxylase and cortisol deficiency (i.e., they are markedly elevated in severe forms and may be normal in patients with the milder forms who are not overtly adrenal insufficient).
#29 3 beta hydroxy steroid dehydrogenase type 1 secreted in other tissues like liver & Placenta is intact. It converts some amount of 17 hydroxy pregnonolone to 17OHP. So there can be high levels of 17OHP as in 21 hydroxylase deficiency.
But this peripherally converted 17OHP is not taken up by adrenals because the circulating concentration is below the Michaelis constant (Km) of P450c21.
#32 In the fetal testis, which normally makes large amounts of testosterone in fetal life, the Leydig cells are destroyed early in gestation, eliminating testosterone biosynthesis; hence, an affected 46,XY fetus does not undergo normal virilization and is born with female external genitalia and a blind vaginal pouch.
However, Wolffian duct derivatives are well developed, indicating the presence of some testosterone synthesis early in fetal life, as predicted by the two-hit model.
The undamaged Sertoli cells produce Mullerian inhibitory hormone, so that the phenotypically female 46,XY fetus has no cervix, uterus, or fallopian tubes.
The fetal ovary makes little or no steroids and contains no steroidogenic enzymes after the first trimester; consequently the ovary remains largely undamaged until it is stimulated by gonadotropins at the time of puberty, when it then produces some estrogen by StAR-independent steroidogenesis.
Because gonadotropin stimulation only recruits individual follicles and does not promote steroidogenesis in the whole ovary, most follicles remain undamaged and available for future cycles.
Although net ovarian steroidogenesis is impaired, enough estrogen is produced (especially in the absence of androgens) to induce breast development, general feminization, monthly estrogen withdrawal, and cyclic vaginal bleeding. However, progesterone synthesis in the latter half of the cycle is disturbed by the accumulating cholesterol esters so that the cycles are anovulatory
#37 Mineralocorticoid therapy in CAH patients returns plasma volume to normal and eliminates the hypovolemic drive to ACTH secretion. Thus, mineralocorticoid therapy often permits the use of lower doses of glucocorticoids in patients with simple virilizing CAH, optimizing growth in children and diminishing unwanted weight gain in adults.
It must be emphasized that a mineralocorticoid is essentially useless unless adequate sodium is presented to the renal tubules
#38 Adrenalectomy has been proposed in severe CAH.
#40 About 20 mg of hydrocortisone has a mineralocorticoid effect of about 0.1 mg of 9a-fluorocortisol
#41 Several studies have established that the secretory rate of cortisol is about 6 to 8 mg/m2 per day. However, effective suppression of ACTH and adrenal androgen production requires somewhat higher doses of about 10 to 15 mg/m2 per day.