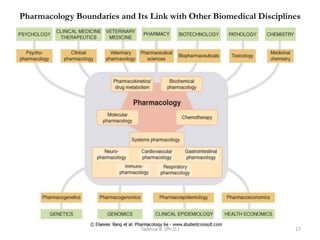
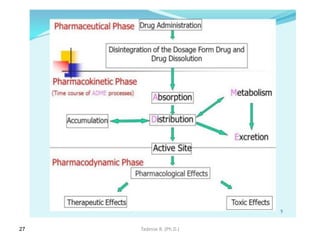
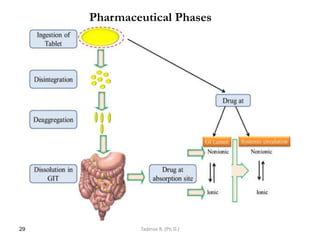
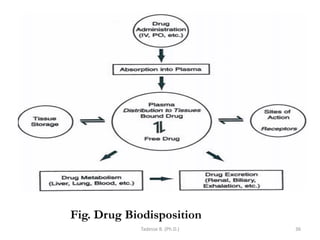
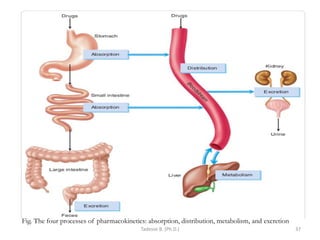
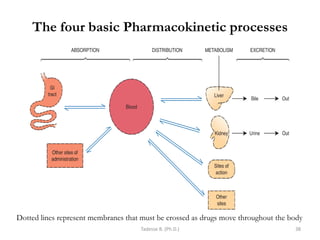
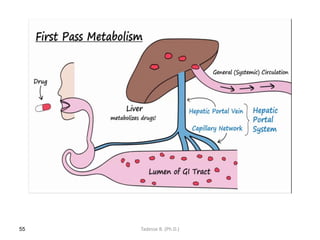
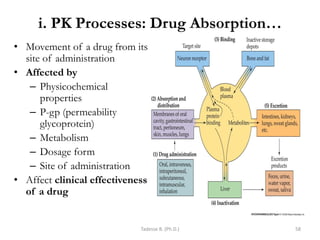
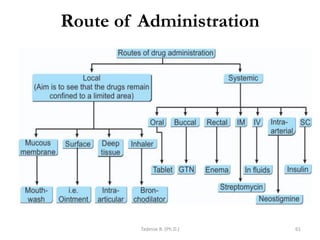
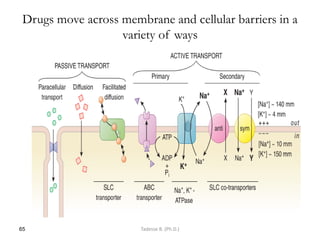
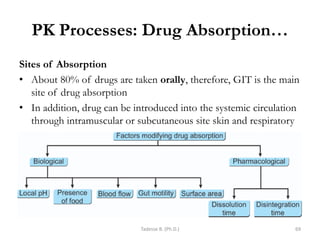
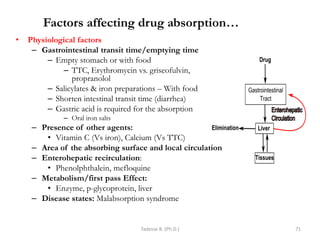
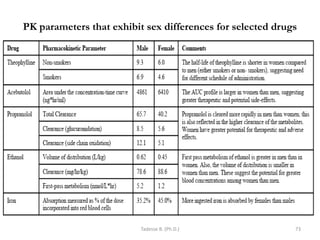
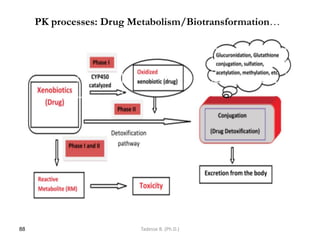
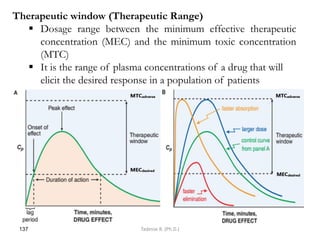
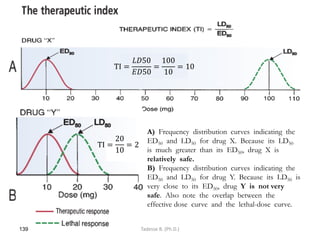
This document provides an overview of the Clinical Pharmacology (Phar521) course for various MSc programs. The course is 3 credit hours and covers topics like pharmacokinetics, pharmacodynamics, drugs used to treat various medical conditions like asthma, diabetes, hypertension, and mental health issues. It also covers analgesics, anesthetics, antimicrobials, and anti-inflammatory drugs. The course contents vary slightly depending on whether the student is in the Maternity and Neonatal Health, Adult Health Nursing, or Pediatric and Child Health Nursing program. The course aims to introduce basic pharmacology principles and applications of pharmacokinetics to clinical practice.

























































![I. Plasma Protein Binding (PPB)
– Drug in systemic circulation exist as bound & unbound form
– Many drugs circulate in the bloodstream bound to plasma
proteins.
• Albumin is a major carrier for acidic drugs;
• α1-acid glycoprotein binds basic drugs such as propranolol.
• Nonspecific binding to other plasma proteins generally occurs to a much
smaller extent.
– Albumin, α1-acid glycoprotein, etc. are the major binding
macromolecules.
– The binding is usually reversible & in dynamic equilibrium
D + P →[DP] → D + P
– As free drugs leave the systemic circulation, the bound drug
dissociate
77
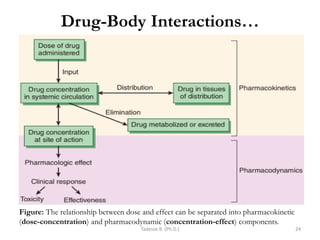
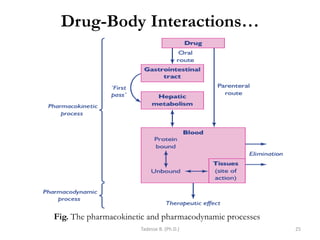
PK processes: Drug Distribution…
Tadesse B. (Ph.D.)](https://image.slidesharecdn.com/chapter1-mscpkspds-ok11-230531072006-8f04118d/85/Chapter-1-MSC-PKs-PDs-Ok11-pdf-77-320.jpg)






















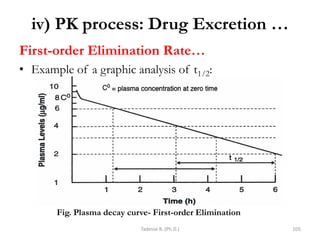


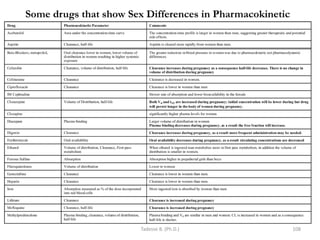




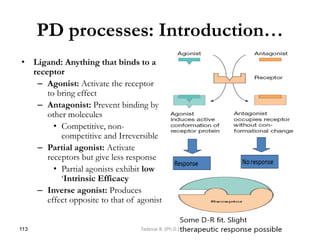

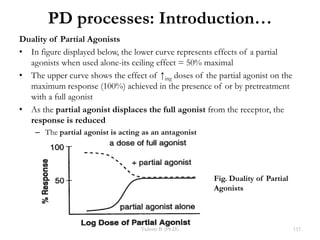










































![General pharmacology modified__ppt[1]](https://cdn.slidesharecdn.com/ss_thumbnails/generalpharmacologymodifiedppt1-210322102730-thumbnail.jpg?width=640&height=640&fit=bounds)


































![Hypothalamus short notes on location, function and disorders by Dr. Neha [PT]...](https://cdn.slidesharecdn.com/ss_thumbnails/hypothalamusbydr-260124142231-2b48143d-thumbnail.jpg?width=640&height=640&fit=bounds)












