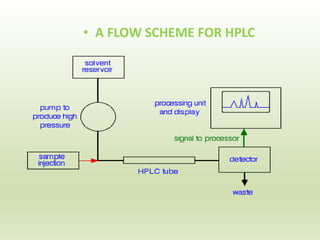
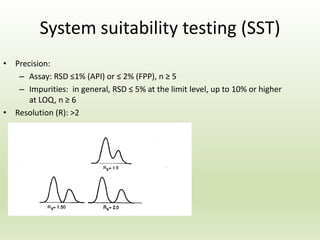
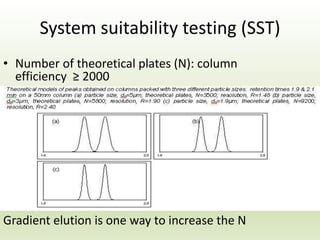
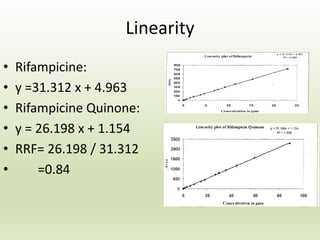
The document describes the development and validation of a reverse phase high performance liquid chromatography (RP-HPLC) method for the estimation of drugs in bulk and pharmaceutical dosage forms. It discusses the need for analytical methods in pharmaceutical analysis and introduces RP-HPLC as a commonly used technique. The document provides details of various studies done to develop and validate RP-HPLC methods for estimating specific drugs and drug combinations in research papers. It also provides chemical and pharmacological profiles of drugs that were estimated using RP-HPLC methods discussed in the literature review section. The aim is to develop a validated RP-HPLC method for estimation of a drug in bulk and marketed dosage form.
![Development and validation of RP-HPLC
method for estimation of bulk dosage form as
marketed dosage form
DHANVANTHRI COLLEGE OF PHARMACEUTICAL SCIENCES
MAHABUBNAGAR, TELANGANA STATE-509002
February-2024 1
C POOJA
M.PHARMACY III SEMESTER
353122885011
[Department of Pharmaceutical Analysis]
2022-2024
Under the guidance of
K SUJATHA, M.Pharm
Assistant Professor](https://image.slidesharecdn.com/cpooja-240330043705-42d3593a/85/ANALYTICAL-METHOD-DEVELOPMENT-AND-VALIDATION-HPLC-UV-1-320.jpg)




![DRUG CATEGORY : Angiotensin receptor blockers
STRUCTURE :
IUPAC NAME : 2-(4{[4-methyl-6-(1-methyl-1H-1,3-benzodiazol-2-yl)-2-
propyl- 1H-1,3-benzodiazol-1-yl]methyl}phenyl)benzoic acid
PHYSICOCHEMICAL PROPERTIES :
Melting point : 261-263 °C
LogP : 3.2
Pka : 4.45
Density : 1.16
Colour : White
Solubility : Soluble in organic solvents
1.5 Drug Profile 8,9,10,11
1. INTRODUCTION
TELMISARTAN](https://image.slidesharecdn.com/cpooja-240330043705-42d3593a/85/ANALYTICAL-METHOD-DEVELOPMENT-AND-VALIDATION-HPLC-UV-7-320.jpg)




























































![01_IJPBA_1739_18[Review]_RA.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/01ijpba173918reviewra-220630064559-6125902f-thumbnail.jpg?width=640&height=640&fit=bounds)


















![Cells and Organs of immune system [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/cellsandorgansofimmunesystemautosaved-260123152717-ea0cb261-thumbnail.jpg?width=640&height=640&fit=bounds)


















