Download to read offline
























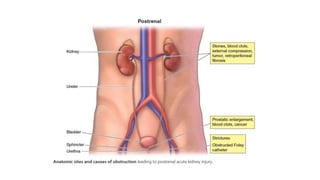




























The document discusses acute kidney injury (AKI) in critically ill patients, including: 1. AKI is common in ICU patients, with various causes like sepsis, ischemia, and nephrotoxins. Proper diagnosis involves assessing fluid status, urinary and serum markers, and biomarkers. 2. Prerenal AKI accounts for most ICU cases and results from reduced renal blood flow due to issues like dehydration. Sepsis is also a major cause through tissue hypoperfusion and endothelial damage. 3. Biomarkers like NGAL and KIM-1 can detect early kidney injury before rises in serum creatinine, but their use is limited by cost currently



























































