PAGE 2
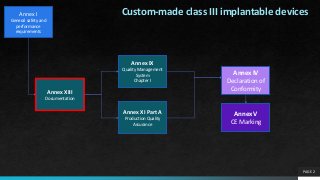
Annex I
Generalsafety and
performance
requirements
Annex XIII
Documentation
Annex XI Part A
Production Quality
Assurance
Custom-made class III implantable devices
Annex IX
Quality Management
System
Chapter I
Annex IV
Declaration of
Conformity
Annex V
CE Marking
3.
PAGE 3
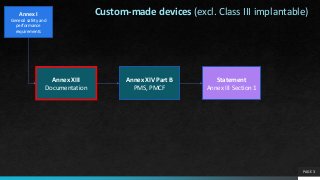
Annex I
Generalsafety and
performance
requirements
Statement
Annex III Section 1
Annex XIII
Documentation
Custom-made devices (excl. Class III implantable)
Annex XIV Part B
PMS, PMCF
PAGE 5
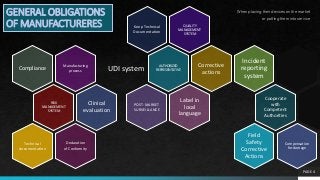
GENERAL OBLIGATIONS
OFMANUFACTURERES
Design and manufacture in compliance with this Regulation
System for risk management (Section 3 of Annex I)
Clinical evaluation, Post-Market Clinical Follow-Up (PMCF) (Article 61 and Annex XIV)
Technical documentation to allow conformity assessment shall be available for 10
years (Annexes II and III)
Technical documentation for custom-made devices (Section 2 of Annex XIII)
Declaration of Conformation, CE marking of conformity (Article 19 and 20)
UDI system (Article 27, 29 and 31)
Technical documentation available for competent authorities for 10 years (implantable
devices for 15 years)
Authorized representative permanently available
List of General Obligations of Manufacturers(Article 10)
6.
PAGE 6
GENERAL OBLIGATIONS
OFMANUFACTURERES
Procedures to ensure conformity with this Regulation
Quality management system
Post-market surveillance system (Article 83).
Label in local language (Section 23 of Annex I)
Obligation to implement corrective actions, withdraw or recall devices in case of
non-conformity
System for recording and reporting incidents and field safety corrective actions
(Articles 87 and 88).
Demonstrate device conformity upon request from Competent authorities
Disclosure of legal or natural persons designated to manufacture devices
Right to claim compensation for damage caused by a defective device
List of General Obligations of Manufacturers(Article 10)
7.
PAGE 7
Statement fora patient or user
Name and address of the manufacturer (all sites)
Name and address of the authorized representative
Data allowing identification of the device
A statement that the device is intended for exclusive use
by a particular identifiable patient or user
Prescribing health professional, incl. health institution
Specific characteristics of the product
Conformity statement
General safety and performance requirements
Medicinal substances incorporated in the device
Keep 10 years (implantable 15 years)
General requirements
Manufacturers shall draw up, keep up to date
and keep documentation available for
conformity assessment
Surveillance
The manufacturer shall review and document
experience gained in the post-production phase,
including Post-Market Clinical Follow-Up and
implement appropriate corrective action
Reporting obligations
• Serious incidents
• Field safety corrective actions
General Obligations of Manufacturers
CUSTOM-MADE DEVICES
List of General Obligations of Manufacturers(Article 10)
8.
DEVICES FOR SPECIALPURPOSES
Member States shall not create obstacles:
Supply of investigational devices to
investigators
Making available custom-made devices
compliant with Annex XIII
Devices w/o CE marking at exhibitions, trade
shows and demonstrations
PAGE 8
9.
Sterile packaging
Indicationhow to recognize sterile packaging
Declaration of sterile condition
Method of sterilization
Manufacturer’s name and address
Description of device
Month and year of manufacture
Time limit for using/implanting the device
Instruction to check integrity of packaging
before use
PAGE 9
“Exclusively for clinical investigations”
“Custom-made device”
10.
Involvement
of Notified
Bodies
Notrequired for devices Class I (non-sterile, non-reusable,
w/o measuring function)
Not required for custom-made devices (excl. custom-made
class III implantable)
Manufacturer may apply to a NB of its choice (one only)
NBs inform each other about withdrawn applications in
EUDAMED
Manufacturer has the duty to disclose withdrawals and
refusals
NB has the right to request any information required for
conformity assessment
NBs are subject to CHAPTER IV and ANNEX VII
Voluntary change of NB shall specify details in an
agreement
PAGE 10
11.
Statement
Manufacturer draws upa Statement:
Name and address of the manufacturer (authorized representative)
Identification of the device
Statement that the device is intended for exclusive use by a particular patient or user
Prescribing professional (and health institution)
Specific characteristics of the product as indicated by the prescription
Statement that the device conforms to Annex I (general safety and performance
requirements)
Information on other components (medicinal substance, human blood or plasma)
PAGE 11
12.
Procedure
Manufacturer shallkeep documentation on the design, manufacture and
performance of the device to allow assessment of conformity
The Statement shall be kept for 10 years (15 years implantable devices)
Section 8 of Annex IX shall apply.
Manufacturer shall review experience from post-production phase (PMCF)
Manufacturer shall report any serious incidents or field safety corrective actions
PAGE 12