Retinitis Pigmetosa_Xerophthalmia_Dr. Pradeep Bastola.pptx
•Download as PPTX, PDF•
0 likes•117 views

Nyctalopia, Retinitis Pigmentosa, Xerophthalmia

Recommended
More Related Content
What's hot
What's hot (20)
Similar to Retinitis Pigmetosa_Xerophthalmia_Dr. Pradeep Bastola.pptx
Similar to Retinitis Pigmetosa_Xerophthalmia_Dr. Pradeep Bastola.pptx (20)
Recently uploaded
Recently uploaded (20)
Retinitis Pigmetosa_Xerophthalmia_Dr. Pradeep Bastola.pptx
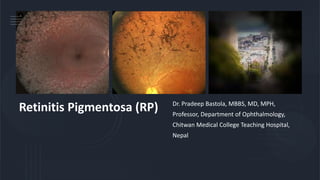
- 1. Retinitis Pigmentosa (RP) Dr. Pradeep Bastola, MBBS, MD, MPH, Professor, Department of Ophthalmology, Chitwan Medical College Teaching Hospital, Nepal
- 2. References • American Academy of Ophthalmology • Wolff’s Ocular Anatomy • Myrin Yanoff • Ryan’s Retina 5th Edition • Clinical Ophthalmology by Jack J. Kanski • The Massachusetts Eye and Ear Infirmary Illustrated Manual of Ophthalmology • AIOS Ready Reckoner in Ophthalmology • Duke Elder Book of Ophthalmology • Various articles from Internet • Presentations of Dr. Pradeep Bastola
- 3. Differential diagnosis of Night Blindness (Nyctalopia) • Xerophthalmia • Retinitis pigmentosa • Primary open angle glaucoma/Advanced glaucoma • High myopia • Post pan-retinal photocoagulation • Congenital rod dystrophy • Congenital stationary night blindness • Familial night blindness • Pharmacologically constricted pupil • Pontine hemorrhage • Opioid poisoning cases • Cuneiform type of cortical cataract
- 4. Retinitis pigmentosa (RP) is a clinically and genetically heterogeneous group of inherited retinal disorders characterized by diffuse progressive dysfunction of predominantly rod photoreceptors with subsequent degeneration of cone photoreceptors and the retinal pigment epithelium (RPE). Visual impairment usually manifests as night blindness and progressive visual field loss. Its prevalence is 1:3000 to 1:5000 RP may be seen in isolation (Typical RP), in association with systemic disease or with atypical presentation (Atypical RP).
- 5. Inheritance: Sporadic (23%) Autosomal recessive (20%) Autosomal dominant (43%) X Linked (8%) Undetermined Inheritance (6%) Prevalence: 1 in 5000, appears in childhood and slowly progresses No racial predilection, M:F::3:2, usually always bilateral Primary retinal dystrophy affecting the rods more than cones and is usually hereditary ICD-10 H35.5
- 6. Risk Factors • There are no known risk factors for RP other than genetic predisposition. • RP may occur as an isolated sporadic disorder or be inherited as autosomal dominant, autosomal recessive, or X-linked. • Mutation of the rhodopsin gene has been reported in many RP cases. • RP is also associated with certain systemic disorders which are usually autosomal recessive. • Many genes associated with RP have been identified and patients can undergo genetic testing. • Association with RPE65 is important as there is now effective gene therapy available for these patients.
- 7. Molecular genetics of RP • More than 100 gene loci that cause RP have been mapped or identified. • Genes that cause RP can be categorized into those that affect the phototransduction cascade, the retinoid cycle, photoreceptor structure, or other biological function of photoreceptor and retinal pigment epithelium. • The most frequent known causes are mutations in the rhodopsin (phototransduction cascade), USH2A (photoreceptor structure), or RPGR (maintenance of cilia or ciliated cells with a possible role in trafficking) genes. • Patients with the same gene defect can have variable severity of disease at a given age. • Despite recent advances, about 50% of cases still have an unknown molecular genetic basis. There is a genetic treatment for RPE65 defects.
- 8. Clinical features Symptoms/Signs Night blindness (Nyctalopia): Diminution of vision in dim light Diminution of vision (Painless gradual, progression depends upon the type of inheritance, AD with best prognosis and AR with worst prognosis) Dark adaption (affected lately) Peripheral visual field loss leading to tubular vision in advanced cases
- 9. Fundus changes (Important) – Triad of pigments, attenuated arterioles and waxy pale optic disc • Retinal pigmentary changes: Typically, perivascular resembling bone corpuscles in shape found in equatorial region but later spread both anteriorly and posteriorly. • Retinal arterioles: Attenuated and later become thread like • Optic disc: Waxy pale but consecutive type of optic atrophy • Presence of Colloid bodies, choroidal sclerosis, cystoid macular edema, atrophic or cellophane maculopathy are also been seen in retina
- 10. Visual field changes • Ring shaped scotoma later the scotoma increase anteriorly and posteriorly and finally a central tubular vision is only left. • Eventually it is also lost, and the patient becomes blind. ERG/EOG: Sub normal eventually extinguished
- 11. Progression of retinitis pigmentosa Fine dust like pigmentation Perivascular ‘bone-spicule’ pigmentation Initially mid-peripheral Unmasking of large choroidal vessels Optic disc pallor Maculopathy Arteriolar attenuation Anterior and peripheral spread
- 12. Associations of retinitis pigmentosa (RP) Ocular: Myopia, primary open angle glaucoma, microphthalmos, conical cornea and posterior sub capsular cataract Systemic: 25% patients have systemic association Laurence Moon Bardet Biedl syndrome: RP, obesity, hypogonadism, polydactyly, mental deficiency Bassen-Kornzweig syndrome: AR, abetalipoproteinemia, RP, diarrhea, Microsomal TG Protein (MTP) Deficiency disease.
- 13. Ocular associations of retinitis pigmentosa Cataract (very common) Keratoconus (uncommon) Optic disc drusen (uncommon) Vitreous degeneration (common) Open-angle glaucoma (uncommon Myopia (common)
- 14. Cockayne’s syndrome: RP, Infantile deafness, dwarfism, mental retardation, nystagmus, ataxia Refsum’s disease: RP, Peripheral neuropathy, cerebellar ataxia Usher’s syndrome: RP and labyrinthine deafness Hallgren’s syndrome: RP, Vestibulo cerebellar ataxia, congenital deafness, mental deficiency Kearns- Sayre Syndrome: RP, AV conduction defects, Mitochondrial transmission, progressive external Ophthalmoplegia, premature death
- 15. Atypical RP • Retinitis pigmetosa sine pigmento: All features of RP but no visible pigments • Sectoral RP: Only a sector of retina involved • Pericentric RP: Pigments around the macula, rest all features present • Retinitis Punctata albescens: Presence of innumerable while dots scattered all over the retina other features include attenuated vessels, night blindness and visual field changes. • Pigmented paravenous chorio-retinal atrophy: Retinal pigments along the veins, usually sporadic, etiology unknown
- 16. Retinitis punctata albescens Presents - usually under 30 years of ages • Scattered white dots extending from posterior pole to periphery • Subsequent development of ‘bone-spicule’ pigmentation Probably variant of retinitis pigmentosa Prognosis - poor ERG - reduced
- 17. Fundus albipunctatus Inheritance - recessive Multitude of tiny yellow-white spots Extend from posterior pole to mid-periphery Congenital stationary night blindness Prognosis - excellent ERG - reduced Fovea spared
- 19. Diagnosis The diagnosis of retinitis pigmentosa is established when the following findings are present. • Bilateral involvement (can be asymmetric) • Impairment of night vision and loss of peripheral vision • Rod dysfunction evidenced by elevated rod final threshold on dark adaptation and/or rod responses on ERG testing that are either reduced in b-wave amplitude and prolonged in implicit time or are essentially non-detectable (extinguished ERG) • Progressive loss in photoreceptor function
- 20. Investigations • Visual fields: Defects leading to tubular vision in advanced RP • Electroretinogram (ERG): Reduction in a and b wave amplitudes • Dark adaptometry: Prolonged in RP • Electrooculogram (EOG): Abnormal in RP • Optical coherence tomography (OCT): Macular edema detection • Fundus fluorescein angiography (FFA): Detects early deterioration of RPE
- 21. Treatment • Vitamin A, Vit E, and Placental extracts to stop progression • Correct refractive error • Acetazolamide, steroids, anti VEGF for cystoid macular edema (CME) • Low vision aid • Rehabilitation • Genetic counselling, and gene therapy • FDA approved Humanitarian Device, called the ARGUS II implant, which may help patients with end-stage RP, only indicated in RP patients with a visual acuity (VA) of perception of light to NPL. CurrentlythereisnocureforRetinitisPigmentosa
- 22. Prognosis • Autosomal dominant RP has the best prognosis, with most patients under 30 years having a visual acuity of 20/30 or better. • X-linked is the most severe form with appreciable impairment of central visual acuity to 20/200 or less by the fifth decade of life. • Autosomal recessive and sporadic cases were intermediate in severity.
- 23. Further reading Boyd K, Vemulakonda GA. Retinitis Pigmentosa. American Academy of Ophthalmology. EyeSmart® Eye health. https://www.aao.org/eye- health/diseases/retinitis-pigmentosa-list. Accessed March 25, 2019.
- 24. XEROPHTHALMIA
- 25. Definition (World Health Organization, 1976) • Xerophthalmia refers to the spectrum of ocular disease caused by severe Vitamin A deficiency (VAD). • Vitamin A serves several essential functions in the eye, and deficiency can lead to a constellation of ocular signs and symptoms that affect the conjunctiva, cornea, and retina. • Xerophthalmia continues to be a leading cause of preventable blindness in developing countries. • Xerophthalmia signifies a severity of VAD that causes significant morbidity and mortality from malnutrition and increased susceptibility to mucosal infections.
- 26. Epidemiology • VAD is among the leading causes of blindness worldwide, estimated to blind half a million children each year • The World Health Organization (WHO) estimated that 228 million children have VAD, causing 1-3 million childhood deaths and 5-10 million cases of eye disease. • VAD is especially prevalent in Africa and South-East Asia, where young children and pregnant women are disproportionally affected
- 27. VAD usually involves a malabsorptive process, such as inflammatory bowel disease or post- gastric bypass surgery, or a severely restrictive diet. VAD is a public Health problem in more than half of all the countries. Vitamin A deficiency affects approximately 21 percent of the developing world's preschool- aged children and leads to the deaths of over 800,000 women and children each year. Prevalence of VAD very low in developed world, in the US (2013), 0.3% According to Nepal Blindness Survey (NBS, 1981), nutritional cause (0.9%) was also a leading cause of blindness, Xerophthalmia was the major nutritional deficiency noted then. Prevalence of VAD in Nepal before Nepal National Vitamin A program was 2-8%.
- 28. Nepal National Vitamin A Program • Nepal Government has initiated National Vitamin A Program (NVAP) in 1993 to improve Vitamin A status of the children aged 6‐59 months and reduce child mortality. • This program has been recognized as a global success story of public health interventions with consistent coverage over 85%. • It initially covered 8 districts and was scaled up nationwide by 2002. • FCHVs distribute the capsules to the targeted children twice a year through a campaign style activity.
- 29. • Biannual deworming tablets distribution: Family Welfare Division, DoHS has been implemented biannual deworming tablets distribution to the children aged 12-59 months aiming to reduce childhood anemia with control or parasitic infestation through public health measures. • This activity is integrated with biannual Vitamin A supplementation to the children aged 6-59 months, which takes place nationally in every ward on first week of Baisakh and Kartik each year.
- 30. Government’s initiations to control of vitamin A deficiency in Nepal • The biannual supplementation of high dose vitamin A capsules to 6–59 months children. • Postpartum vitamin A supplementation for mothers within 42 days of delivery. • Strengthen implementation of vitamin A treatment protocol for severe malnutrition, persistent diarrhoea, measles and xerophthalmia. • Nutrition education to promote dietary diversification and consumption of vitamin A rich foods. • Ensuring the availability of vitamin, A capsules at health facilities. • Increase awareness of importance of vitamin A supplementation. • The biannual distribution of vitamin A capsules to 6- to 59 months children through FCHVs. • Advocate for increased home production, consumption and preservation of vitamin A rich foods. • Strengthen the use of the vitamin A Treatment protocol.
- 31. Etiology of Xerophthalmia (VAD) Reduced intake of Vitamin A Impaired absorption of Vitamin A Reduced Storage of Vitamin A • Inadequate food supply • Chronic alcoholism • Highly selective dieting • Dysphagia • Mental illness • Crohn's disease • Celiac disease • Pancreatic insufficiency • Short bowel syndrome • Chronic diarrhea • Inflammatory bowel disease • Upper gastrointestinal surgery • Giardiasis • Abetalipoproteinemia • Liver disease • Cystic fibrosis
- 32. Recommended daily intake of Vitamin A • The recommended dietary allowance of vitamin A is 700ug/day in females and 900ug/day in the males. • For children and pregnant or lactating women, the recommended amount is 300-900, 770, and 1300ug/day, respectively. • Children aged 1-5 years old require a minimum of 200ug/day to prevent symptomatic VAD. • Fat soluble Vitamin, 80-90% stored in the Liver Dietary sources of preformed vitamin A include dark leafy greens, orange-colored vegetables, fish liver oils, liver, egg yolks, butter, and vitamin A-fortified dairy products.
- 33. Risk factors • Living in an endemic area • Low socioeconomic status • Malnutrition • Maternal malnourishment (affects Vitamin A concentration in breastmilk) • Zinc deficiency: inadequate zinc can depress the hepatic synthesis of retinol-binding protein (RBP), which is required for metabolism of retinol from the liver. Zinc may also play a role in the conversion of beta-carotene to retinol via the enzyme 15-15’-dioxygenase. • Co-existing measles or other respiratory/diarrheal illness • Vitamin A has several essential functions in the body, including cell development, metabolism, immune function, vision, and reproductive function.
- 34. Diagnosis (WHO Classification of Xerophthalmia, 1976 based on clinical signs) Grade of xerophthalmia Peak age group (years) Type of deficiency XN: Night blindness 2-6; adult women Longstanding. Not blinding X1A: Conjunctival xerosis 3-6 Longstanding. Not blinding X1B: Bitot's spots 3-6 Longstanding. Not blinding X2: Corneal xerosis 1-4 Acute deficiency. Can be blinding X3A: Corneal ulcer <1/3 cornea 1-4 Severe acute deficiency. Blinding X3B: Corneal ulcer/keratomalacia1/3 cornea or greater 1-4 Severe acute deficiency. Blinding XS: Corneal scarring (from X3) >2 Consequence of corneal ulceration XF: Xerophthalmos fundus Adults Longstanding. Not blinding. Rare
- 35. Night blindness (Grade XN): Defective vision in dim light or night blindness is one of the most common manifestations of VAD, especially in children aged 2-6 years or pregnant or lactating women. Although it is considered one of the earliest manifestations, children with VAD may develop one of the more severe signs, such as corneal ulcers, after infection or diarrhoea without any of the classically early signs. Children may not be able to verbalize their symptoms, and parents need to be asked if they have noticed their children behaving differently in the dark, e.g., becoming less active or fearful. Night blindness is both a sensitive and specific indicator for serum retinol levels. Conjunctival xerosis (Grade X1A): Conjunctival xerosis is characterized by a dull and dry appearance of the conjunctiva with slight wrinkling. It is caused by the loss of goblet cells and insufficient mucin secretion, and it can be subtle and difficult to detect clinically.
- 36. Bitot spots (Grade X1B): Bitot spots are collections of desquamated, keratinized epithelial cells mixed with the gas-forming bacteria Corynebacterium xerosis. • They appear as triangular patches of foamy, whitish, opaque deposits, typically located on the bulbar conjunctiva near the limbus at the 3 and 9 o’clock positions. They are more common temporally. Corneal xerosis (Grade X2): Conjunctival xerosis is characterized by a dull and hazy appearance of the cornea that is caused by drying of the cornea secondary to conjunctival gland dysfunction. • It may initially present with bilateral punctate corneal epithelial erosions, and it can quickly progress to the stage of corneal ulceration. • Up to this stage, high-dose Vitamin A supplementation can result in the full preservation of vision.
- 37. Corneal ulceration (Grade X3A and B): Corneal xerosis can lead corneal ulceration and melting if not treated urgently. Keratomalacia, the melting away of the cornea by liquefactive necrosis, is the most severe form of xerophthalmia, which can perforate and destroy the cornea in just a matter of days. A child who appears relatively healthy but develops keratomalacia should be questioned about a recent history of measles or diarrhea, which could rapidly deplete already deficient vitamin A stores. Corneal scar (Grade XS): Corneal scarring due to VAD is often symmetric and bilateral. Other causes of corneal scarring must be ruled out.
- 38. Xerophthalmic fundus (Grade XF): Prolonged VAD can lead to structural changes in the retina. Fundus changes appear as small, white, deep retinal lesions scattered throughout the posterior pole.
- 39. Work up/Investigations • History, and physical examination • Clinical signs • Serum vitamin A/retinol (reference range: 20-60 mcg/dL). (VAD - related ocular symptoms have been shown to develop at concentrations <10mcg/dL) • Serum retinol binding protein (reference range: 30-75 ug/ml). • Serum zinc (reference range: 75-120 mcg/dL)
- 40. • Dark adaptometry and night vision threshold tests - Prolonged • Electroretinogram (ERG): Retinopathy from VAD is associated with decreased amplitude • Impression cytology: Conjunctival specimens can be viewed for the presence of goblet cells, a decrease in normal amount is considered an effective measurement of VAD • Liver biopsy: Considered the gold standard for evaluating total body vitamin A, not practiced except for research
- 41. Differential Diagnosis • Parasitic eye disease, e.g., Acanthamoeba keratitis or Onchocerciasis • Trachoma • Allergic conjunctivitis • Viral conjunctivitis • Dry eye syndrome • Retinitis pigmentosa and other retinal dystrophies that can present with night blindness • Pinguecula/pterygium (may resemble Bitot spots)
- 42. Treatment (Oral or Intramuscular Vitamin A supplement) Keratomalacia should be treated as a medical emergency, as it is an indicator of very severe VAD Vitamin A Dosage International Units (IU), (IM dose half of oral dose) Young infants 0-5 months1 50,000 Older infants 6-11 months (less than 8kgs)1 100,000 Children (males: 12 months or more; females 12 months to 12 year and 50 y or more)1 200,000 Women (13-49 y) with night blindness and/or Bitot's spots 10,000 every day or 25,000 every week for at least 3 mo Women (13-49 y) with active corneal lesions 200,000 on days 1, 2, and 14 Vitamin A treatment regimens. 1 Schedule: severe malnutrition, day 1; measles, days 1 and 2; xerophthalmia, days 1, 2, and 14.
- 43. Patients with concomitant zinc deficiency should also undergo zinc supplementation. Keratomalacia in Xerophthalmia is treated in line with the Bacterial Corneal Ulcer. VAD associated with malabsorptive, or other processes is treated based on the severity of deficiency and provider discretion, usually involving daily treatment. Localized ocular treatment includes intense lubrication, topical retinoid acid, and management of perforation. Patient education is a large component of management, and patients and their families should be educated on dietary sources of vitamin A and well-balanced, nutrient-rich diets. Alcohol abuse should be addressed if applicable. In resource poor countries, large-scale vitamin A supplementation programs are in place eg, Nepal, India, Sub Saharan Africa
- 44. Prognosis Nearly 2/3 of children with keratomalacia die within months. Mortality in children with night blindness is triple the mortality found in children with subclinical VAD, and mortality in children with both Bitot spots and night blindness is reported to be nine times that of children with subclinical VAD. With prompt treatment of high-dose vitamin A, the early Ophthalmologic signs, such as a rod function, conjunctival xerosis, and night blindness, can resolve completely within about 2 months of supplementation, without long-term sequelae. Corneal xerosis and ulceration may also improve; however, they can lead to scarring and permanent vision loss.