1. Treatment effect on diversity and evolution
2,470 sequences from 26 patients over 290 time points were
analyzed. The results showed a slight increase in nucleotide
diversity among gp120 and gp41, and a decrease in diversity
among protease over time. The average rate of change for gp120
and gp41 was 0.15% per month (~5.5bp/mo), reverse
transcriptase was 0.04% per month (~0.8bp/mo), while the
protease gene was -0.02% per month (~0.5bp/mo).
Predicting treatment from protease sequence
16,075 sequences for the protease gene from patients with
known treatment status were obtained. 10,923 (68%) were from
patients with a history of treatment. Multiple SVM kernels were
evaluated based on sensitivity and specificity (respectively): radial
(0.92, 0.90), polynomial (0.92, 0.85), linear (0.92, 0.84), and
hyperbolic tangent (1, 0). Features were iteratively removed,
finding that performance was maintained until 10 nucleotides
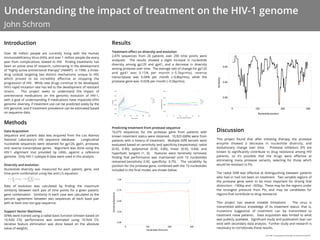
remained (sensitivity: 0.92, specificity: 0.75). The variability by
position for the protease gene, combined with the 10 nucleotides
included in the final model, are shown below.
John Schrom
Understanding the impact of treatment on the HIV-1 genome
Introduction
Methods
Results
Discussion
Over 36 million people are currently living with the Human
Immunodeficiency Virus (HIV), and over 1 million people die every
year from complications related to HIV. Finding treatments has
been an active area of research, culminating in the development
of “highly active antiretroviral therapy” (HAART) in 1996: a three-
drug cocktail targeting two distinct mechanisms unique to HIV,
which proved to be incredibly effective at stopping the
progression of HIV. While new drugs continue to be developed,
HIV’s rapid mutation rate has led to the development of resistant
strains. This project seeks to understand the impact of
antiretroviral medications on the genomic evolution of HIV-1,
with a goal of understanding if medications have impacted HIV’s
genomic diversity, if treatment use can be predicted solely by the
HIV genome, and if treatment prevalence can be estimated based
on sequence data.
This project found that after initiating therapy, the protease
enzyme showed a decrease in nucleotide diversity, and
evolutionary change over time. Protease inhibitors (PI) are
known to significantly contribute to drug resistance among HIV
patients, so it’s possible that the drugs were effective at
eliminating many protease variants, selecting for those which
would be resistant to PIs.
The radial SVM was effective at distinguishing between patients
who had or had not been on treatment. Two variable regions of
the protease gene seem to be most important for driving that
distinction: ~180bp and ~265bp. These may be the regions under
the strongest pressure from PIs, and may be candidates for
regions that contribute to drug resistance.
This project has several notable limitations. The virus is
transmitted without knowledge of its treatment status: that is,
mutations suggestive of treatment can be transmitted to
treatment naive patients. Data acquisition was limited to what
was publicly available. Significant study and publication bias can
exist with secondary data analysis. Further study and research is
necessary to corroborate these results.
Data Acquisition
Sequence and patient data was acquired from the Los Alamos
National Laboratory’s HIV sequence database. Longitudinal
nucleotide sequences were obtained for gp120, gp41, protease,
and reverse transcriptase genes. Alignment was done using the
HIV-1 alignment tool provided by LANL, against a reference
genome. Only HIV-1 subtype B data were used in this analysis.
Diversity and evolution
Nucleotide diversity was measured for each patient, gene, and
time point combination using Nei and Li’s equation:
Rate of evolution was calculated by finding the maximum
similarity between each pair of time points for a given patient,
gene combination. Similarity in each case was calculated as the
percent agreement between two sequences at each base pair
with at least one non-gap sequence.
Support Vector Machines
SVMs were trained using a radial basis function (chosen based on
10-fold CV); performance was estimated using 10-fold CV;
iterative feature elimination was done based on the absolute
value of weights.
CSCI 5481: Computational Techniques for Genomics; Fall 2015