
Recommended
More Related Content
What's hot
What's hot (20)
Similar to US Medical Research And Dvt
Similar to US Medical Research And Dvt (20)
US Medical Research And Dvt
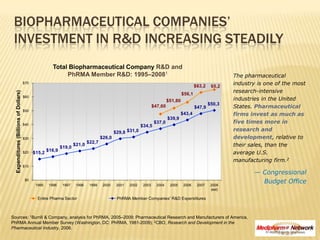
- 1. Biopharmaceutical Companies’ Investment in R&D Increasing Steadily Total Biopharmaceutical Company R&D and PhRMA Member R&D: 1995–20081 The pharmaceutical industry is one of the most research-intensive industries in the United States. Pharmaceutical firms invest as much asfive times more in research and development, relative to their sales, than the average U.S. manufacturing firm.2 — Congressional Budget Office Sources: 1Burrill & Company, analysis for PhRMA, 2005–2009; Pharmaceutical Research and Manufacturers of America, PhRMA Annual Member Survey (Washington, DC: PhRMA, 1981-2009); 2CBO, Research and Development in the Pharmaceutical Industry, 2006.
- 2. Clinical Research Clinical Research Translational Research Translational Research Basic Research Basic Research Federal and Industry Roles in Research and Development Government and biopharmaceutical industry research are complementary Private Sector – $65.2B1 There is an ecosystem of science and biotechnology. Public organizations, patient organizations, universities, Congress, FDA, all of this is an ecosystem that is envied in the rest of the world. – E. Zerhouni, Director of NIH NIH3– $29.4B total – $20.1B research Sources: 1Burrill & Company, analysis for PhRMA, 2005–2009 (Includes PhRMA research associates and nonmembers) in PhRMA, “Profile 2008, Pharmaceutical Industry;” PhRMA, “PhRMA Annual Membership Survey,” 1996-2009; 2Adapted from E. Zerhouni, Presentation at Transforming Health: Fulfilling the Promise of Research, 2007; 3NIH Office of the Budget, “FY 2009 President’s Budget Request Tabular Data”, http://officeofbudget.od.nih.gov/ui/2008/tabular%20data.pdf
- 3. Medical Research in the U.S. Outpaces the Rest of the World …in the late 1980s only 41% of the top 50 innovative drugs were of American origin, in the late 1990s…[it had] climbed to 62%.... In 1990, the pharmaceutical industry spent 50% more on research in Europe than in the U.S. In 2001, the situation was reversed with 40% more spent in the U.S.2 –Gunter Verheugen, Vice-President of the European Commission for Enterprise and Industry Number of Compounds in Development, by Region,* 1997–2007 *Note: Reflects the number of compounds in clinical trials or awaiting approval as of June of each year. Compounds in development for multiple regions are counted in each region for which regulatory approval is sought, and multiple indications are counted only once. Sources: 1Adis R&D Insight, February 2009; 2G. Verheugen, “Address to the Concluding Session of the European Track” (Lyon) 2005.
- 4. Post-MarketingSurveillance Drug Discovery Preclinical Clinical Trials FDA Review Scale-Up to Mfg. 5 250 ~ 5,000 – 10,000 COMPOUNDS ONE FDA-APPROVED DRUG PRE-DISCOVERY PHASE 3 PHASE 1 PHASE 2 NDA SUBMITTED IND SUBMITTED NUMBER OF VOLUNTEERS 20–100 100–500 1,000–5,000 0.5 – 2 YEARS INDEFINITE 6 – 7 YEARS 3 – 6 YEARS Drug Development takes longer that it did in the past Developing a new medicine takes an average of 10–15 years; the Congressional Budget Office reports that “relatively few drugs survive the clinical trial process” Sources: Drug Discovery and Development: Understanding the R&D Process, www.innovation.org; CBO, Research and Development in the Pharmaceutical Industry, 2006.
- 5. The Cost of Developing a New Drug Has Greatly Increased Cost to Develop One New Drug1 Billions (Constant Dollars, Year 2000) Sources: 1J. DiMasi and H. Grabowski, "The Cost of Biopharmaceutical R&D: Is Biotech Different?," Managerial and Decision Economics, 2007; J. DiMasi et al., “The Price of Innovation: New Estimates of Drug Development Costs,” Journal of Health Economics, 2003.
- 6. Increasing Complexity of Clinical Trials During the last decade clinical trial designs andprocedures have become much more complex, demanding more staff time and effort, and discouraging patient-enrollment and retention Definitions: Procedures: include lab & blood work, routine exams, x-rays & imaging, questionnaire & subjective assessments, invasive procedures, heart assessment, etc. Protocol: the clinical-trial design plan Enrollment rate: the percentage of volunteers meeting the increasing number of protocol eligibility criteria (percentage screened who were then enrolled) Retention rates: the percentage of volunteers enrolled who then completed the study; declining retention rates mean that firms must enroll more patients initially and/or recruit more patients during the trial. Source: Tufts Center for the Study of Drug Development, “Growing Protocol Design Complexity Stresses Investigators, Volunteers,” Impact Report, 2008.
- 7. Probability of Success for Investigational Drugs Is Small Approximately 20% of self-originated new drugs that enter clinical testing will receive U.S. marketing approval.1 Clinical Approval Success Rates by Therapeutic Class1 Source: 1Tufts Center for the Study of Drug Development, “New drugs entering clinical testing in top 10 firms jumped 52% in 2003-05,” Impact Report, 2006.
- 8. Cumulative Approvals for Medicines: 1990–2008 Many examples exist of major therapeutic gains achieved by the industry in recent years… anecdotal and statistical evidence suggests that therapid increases that have been observed in drug-related R&D spending have been accompanied bymajor therapeutic gains in available drug treatments.2 —Congressional Budget Office Number of New Drug Approvals Note: New medicines include New Drug Applications and Biologics License Applications Sources: 1Food and Drug Administration, www.fda.gov; PhRMA, “New Drug Approvals,” Reports 1991-2008; “New Molecular Entities Approved in 2008,” The Pink Sheet, January 2009; 2CBO, Research and Development in the Pharmaceutical Industry, 2006.
- 9. Even After Approval, Few Medicines Are a Commercial Success Lifetime Sales Compared to Average R&D Costs After-Tax Present Value of Sales (Millions of 2000 Dollars) New Rx Drugs Introduced Between 1990 and 1994, Grouped by Tenths, by Lifetime Sales Note: Drug development costs represent after-tax out-of-pocket costs in 2000 dollars for drugs introduced from 1990–94. The same analysis found that the total cost of developing a new drug was $1.3 billion in 2006. Average R&D Costs include the cost of the approved medicines as well as those that fail to reach approval. Sources: J. Vernon et al., “Drug Development Costs when Financial Risk is Measured Using the Fama-French Three Factor Model,” Unpublished Working Paper, 2008; J. DiMasi and H. Grabowski, “The Cost of Biopharmaceutical R&D: Is Biotech Different?,” Managerial and Decision Economics, 2007.
- 10. Importance of Post-Approval R&D 2003 Research into new uses for approved biologics is an important source of medical advances 1998 For use in combination with Rebetol to treat pediatric hepatitis C.2 As of 2007, “47%of BLAs for recombinant DNA products and monoclonal antibodies regulated by CDER ha[d] at least one additional FDA-approved indication after the initial approval…. One-third of all additional indications of biotechnology-derived protein drugs studied were approved by the FDA more than seven years after approval of the initial indication. —Boston Consulting Group 1997 For use in combination with Rebetol capsules for chronic viral hepatitis C in patients with compensated liver disease who have relapsed following alfa Interferon therapy. To treat hepatitis B in pediatric patients.2 For use in combination with Rebetold capsules for chronic viral hepatitis C in patients with compensated liver disease previously untreated with alfa Interferon therapy. 1995 For treatment of clinically aggressive follicular non-Hodgkin’s lymphoma in conjunction with antracycline-containing combination therapy. 1993 For use adjuvant to surgical treatment for malignant melanoma for those patients free of disease but at high risk for systemic recurrence within 56 days of surgery. 1992 For use in condylomata acuminata with podophyllin. 1991 For treatment of hepatitis B in patients 18 years or older with compen-sated liver disease. 1Orphan Indication 2Pediatric Indication 1988 To treat patients with chronic hepatitis non-A, non-B/C with compensated liver diseases and history of blood or blood product exposure and/or who are HCV anti-body positive. For intralesional treatment of select cases of condylomata acuminata involving external surfaces of genital and perianal areas. For treatment of select patients with AIDS-related Kaposi’s sarcoma¹ 1986 Example: Intron-A (interferon-alfa-2b) FDA Approval of Original Indication: To treat hairy cell leukemia Sources: Boston Consulting Group, “Continued Development of Approved Biological Drugs,” White Paper, 2007; PhRMA, “Post-Approval Research on Biotech Medicines Leads to Key Medical Advances,” PhRMA Report, 2007.