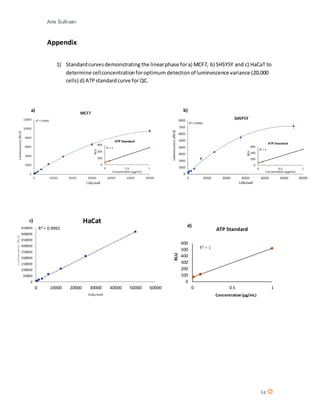
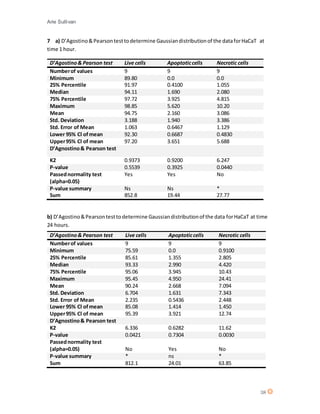
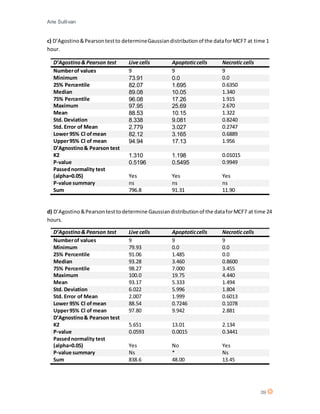
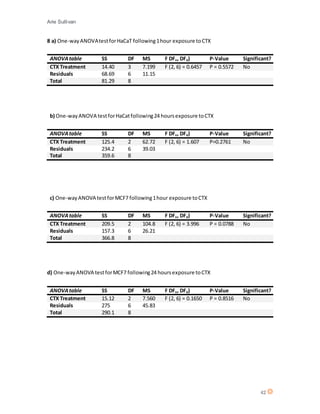
This document discusses the mechanism of action of Chlorotoxin (CTX), a peptide derived from scorpion venom. It summarizes that CTX shows specificity for binding to certain tumors but the exact mechanism is unclear, with three potential receptors identified: chloride channels, matrix metalloproteinase-2 (MMP-2), and annexin A2. The document also reviews the potential of venoms as therapeutics and discusses CTX's ability to "paint" tumors, making them visible for surgeons. Experiments are described that aimed to determine CTX's effects on various cell lines, including ones of neuroectodermal origin, and investigate whether it induces apoptosis or a new mechanism of necrosis.