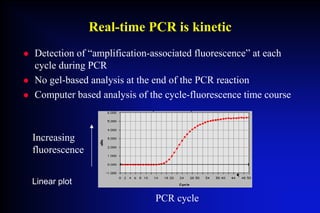
This document provides an overview of real-time and quantitative PCR (RTAQ-PCR) technology. It discusses how RTAQ-PCR uses fluorescence detection during PCR cycles to quantify targets in real-time, rather than relying on end-point analysis. Software is used to analyze amplification curves and calculate threshold cycle values, which correlate inversely with starting target amounts. Both non-specific dye-based and specific probe-based fluorescence detection methods are described. The document also reviews considerations for assay design and quantitative analysis methods like absolute and relative quantification.