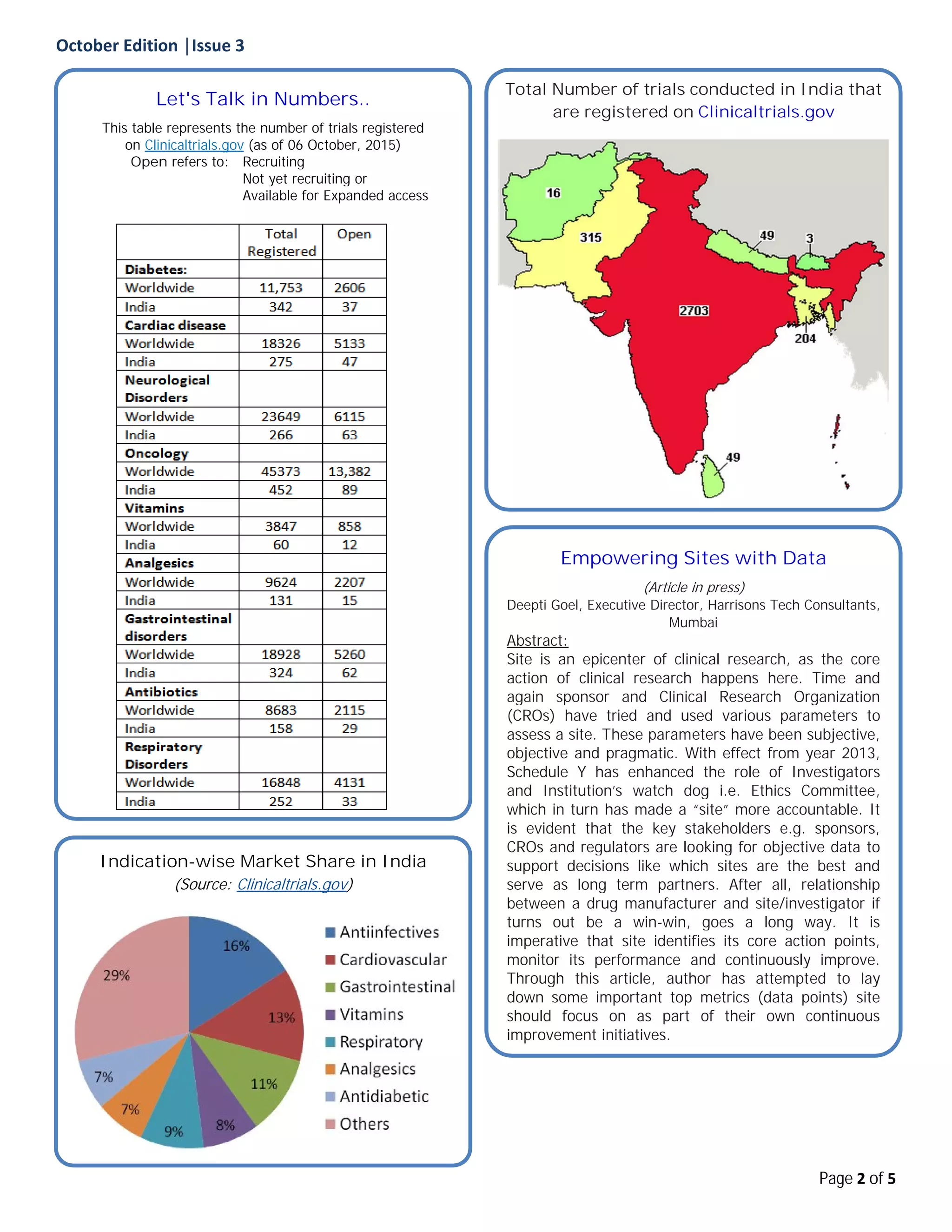
The document provides an overview of clinical trial statistics and regulatory updates from India. It discusses the increasing number of registered ethics committees in India and importance of quality and compliance. The newsletter also highlights key metrics sites should focus on to improve performance and notes recent Indian regulatory milestones related to clinical trials. Additionally, it briefly summarizes an FDA approval of a new drug and a platform aiming to improve patient recruitment for clinical trials.