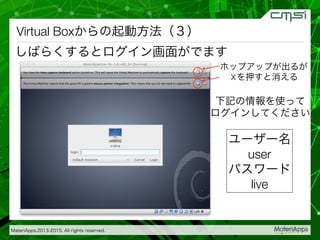
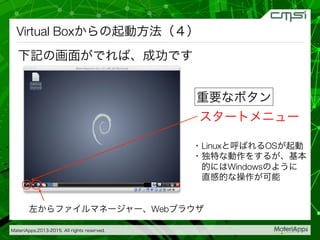
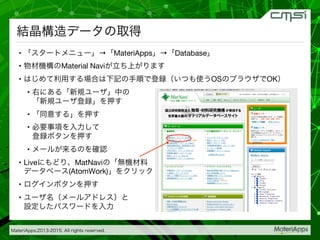
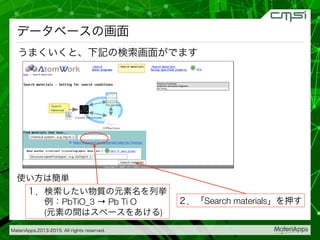
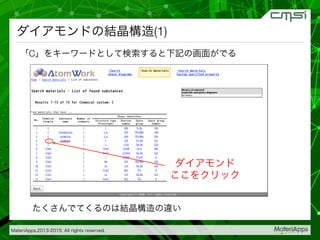
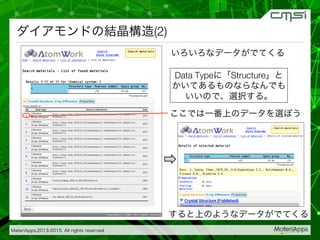
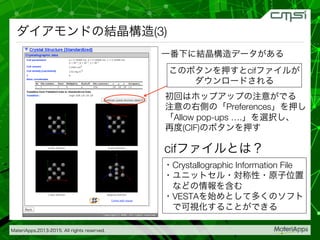
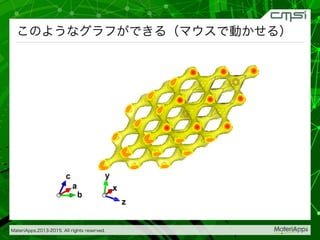
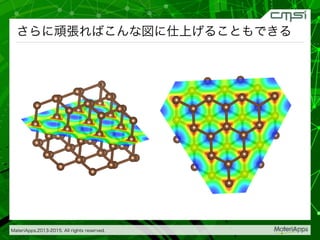
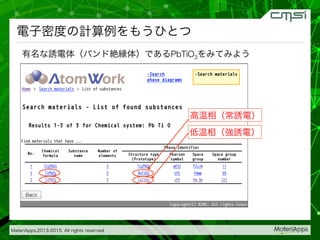
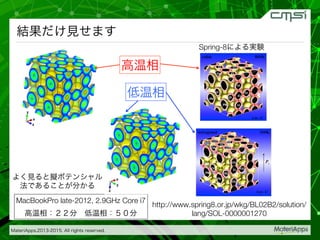
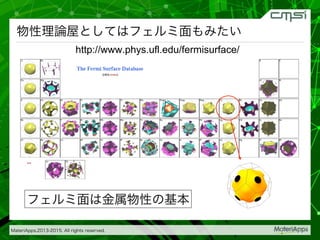
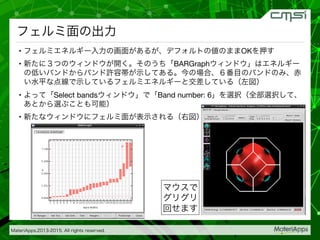
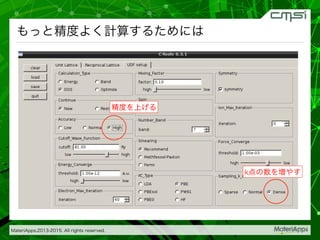
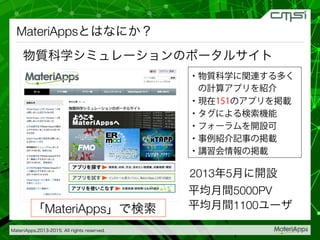
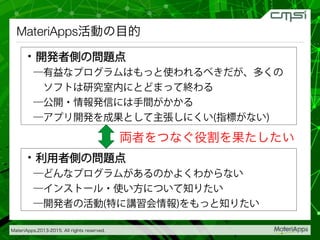
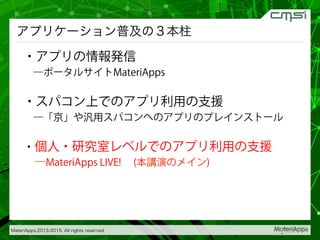
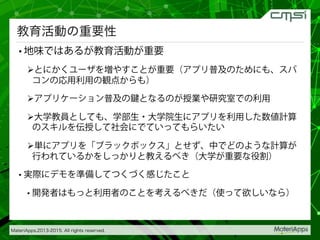
2015-07-28@SPring-8材料構造の解析に役立つ計算科学研究会 • MateriApps LIVE!の起動方法の説明 • 第一原理計算手法によるバンド計算の実演 • 分子動力学による溶液のシュミレーション