2. BIOPHARMACEUTICAL FACTORS IN DOSAGE FORM DESIGN
OR
FACTORS INFLUENCING GI ABSORPTION OF DRUG
PHARMACEUTICAL FACTOR
PATIENT RELATED FACTORS
3. PHARMACEUTICAL FACTOR
Physicochemical properties of drug
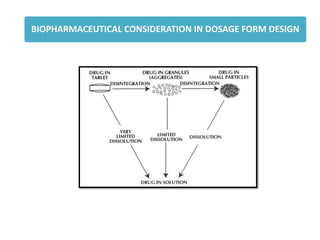
Dosage form, dosage form related,
pharmaceutical excipient
(Pharmaco Technical Factor)
1. Drug solubility and dissolution
rate
2. Particle size and effective surface
area
3. Polymorphism and amorphism
4. Pseudopolymorphism
(hydrates/solvates)
5. Salt form of the drug
6. Lipophilicity of the drug
7. pKa of the drug and
gastrointestinal pH
8. Drug stability
9. Stereochemical nature of the
drug
1. Disintegration time
(tablets/capsules)
2. Dissolution time
3. Manufacturing variables
4. Pharmaceutical ingredients
(excipients/adjuvants)
5. Nature and type of dosage form
6. Product age and storage conditions
4. Physicochemical properties of drug
Particle size and effective surface area
• Solubility, the phenomenon of dissolution of solute in solvent to give a
homogenous system.
• The process by which a solid or liquid forms a homogeneous mixture with a
solvent or mass transfer form solid phase to liquid phase
5. Particle size and surface area of a solid drug are inversely related to each other. Smaller the
drug particle, greater the surface area.
Absolute surface area Effective surface area
Micronisation has in fact enabled the formulator to decrease the dose of
certain drugs because of increased absorption.
GRISEOFULVIN dose was reduced to half and that of SPIRONOLACTONE was
decreased 20 times following micronisation.
For lipophilic drugs ASPIRIN, PHENACETIN AND PHENOBARBITAL, micronisation
actually results in a decrease in the effective surface area. It leads to fall in dissolution
rate.
6. Particle Size Reduction
The absolute surface area of hydrophobic drugs can be converted to their
effective surface area by:
1. Use of surfactant as a wetting agent that - Decreases the interfacial tension, and
Displaces the adsorbed air with the solvent.
For example, polysorbate 80 increases the bioavailability of phenacetin by promoting its
wettability.
2. Adding hydrophilic diluents such as PEG, PVP, dextrose, etc. which coat the surface of
hydrophobic drug particles and render them hydrophilic.
7. Particle size reduction and subsequent increase in the surface area and dissolution rate is
not advisable under following circumstances –
When the drugs are unstable and degrade in solution form (penicillin G and
erythromycin),
When drugs produce undesirable effects (gastric irritation caused by nitrofurantoin)
When a sustained effect is desired.
Reducing the particle size to a submicron to improve dissolution through solubility is
possible by use of one techniques such as formation of:
1. Molecular dispersion/solid solution where the sparingly soluble drug is molecularly
entrapped in the lattice of a hydrophilic agent such as cyclodextrins.
2. Solid dispersion where the drug is dispersed in a soluble carrier such as
PVP, PEG, urea, etc.
9. STABLE POLYMORPH
Represents the lowest energy state, has highest melting point and least aqueous
solubility.
METASTABLE FORMS
Represent the higher energy state, have lower melting points and higher aqueous
solubilities.
A is the stable polymorph
B is the Metastable polymorph
(more soluble)
C is the unstable polymorph
10. AMORPHOUS FORM
Amorphous form having no internal crystal structure.
Highest energy state and can be considered as supercooled liquids.
They have greater aqueous solubility than the crystalline forms
[as the energy required to transfer a molecule from crystal lattice is greater than
that required for non-crystalline (amorphous) Solid].
Example
1. Amorphous form of novobiocin is 10 times more soluble than the crystalline
form.
2. Chloramphenicol palmitate, cortisone acetate and phenobarbital are other
examples where the amorphous forms exhibit higher water solubility.
Amorphous > Metastable > Stable
11.
12. Hydrates and Solvate
The stoichiometric type of adducts where the solvent molecules are
incorporated in the crystal lattice of the solid are called as the solvates, and the
trapped solvent as solvent of crystallization.
The solvates can exist in different crystalline forms called as pseudopolymorphs.
This phenomenon is called as pseudopolymorphism.
When the solvent in association with the drug is water, the solvate is known as a
hydrate. Hydrates are most common solvate forms of drugs.
The anhydrous form of a drug has greater aqueous solubility than the hydrates.
The anhydrous form of theophylline and ampicillin have higher aqueous
solubilities, dissolve at a faster rate and show better bioavailability in
comparison to their monohydrate and trihydrate forms respectively.
13. The organic (nonaqueous) solvates have greater aqueous solubility than the
non-solvates—for example,
n-pentanol solvate of fludrocortisone and succinylsulphathiazole
chloroform solvate of griseofulvin are more water-soluble than their non-
solvated forms.
14. SALT FORM OF DRUG
Dissolution process of a salt form of a weakly acidic drug in gastric fluid.
15. - One example is the dissolution and bioavailability profiles of Benzathine
and various salts.
Choline and the isopropanolamine salts of theophylline dissolve 3 to 4 times more
rapidly than the ethylenediamine salt and show better bioavailability.
Smaller the size of the counter ion, greater the solubility of salt
Bioavailability of novobiocin from its sodium salt, calcium salt and free acid form
was found to be in the ratio — 50 : 25 : 1.
16. Drug pKa and Lipophilicity and GI pH—pH Partition Hypothesis
- According to the pH-partition hypothesis, the gastrointestinal
epithelia acts as a lipid barrier towards drugs which are
absorbed by passive diffusion, and those that are lipid soluble
will pass across the barrier.
Brodie et al.
“The theory states that for drug compounds of molecular weight
greater than 100, which are primarily transported across the
biomembrane by passive diffusion, the process of absorption is
governed by:
dissociation constant (pKa) of the drug,
lipid solubility of the unionised drug (a function of drug Ko/w)
and The pH at the absorption site.
3/6/2024
17. Most drugs are weak electrolytes (weak acids or weak bases), their degree of
ionisation depends upon the pH of the biological fluid.
If the pH on either side on the membrane is different, then the compartment whose
pH favours greater ionisation of the drug will contain greater amount of drug, and
only the unionised or undissociated fraction of drug, if sufficiently lipid soluble, can
permeate the membrane passively until the concentration of unionised drug on
either side of the membrane becomes equal i.e. until equilibrium is attained.
The above statement of the hypothesis was based on the assumptions that:
1. The GIT is a simple lipoidal barrier to the transport of drug.
2. Larger the fraction of unionised drug, faster the absorption.
3. Greater the lipophilicity (Ko/w) of the unionised drug, better the absorption.
3/6/2024
18. Drug pKa and Gastrointestinal pH
The lower the pKa of an acidic drug, stronger the acid
i.e. greater the proportion of ionised form at a particular pH.
Higher the pKa of a basic drug, stronger the base
i.e. greater the proportion of ionised form at a particular pH.
The knowledge of pKa of drug and pH at the absorption site (or biological fluid), the
relative amount of ionised and unionised drug in solution at a particular pH and the
percent of drug ionised at this pH can be determined by Henderson-Hasselbach
equations:
3/6/2024
19. WEAK ACIDS
WEAK BASE
If there is a membrane barrier that separates the aqueous solutions of different pH such
as the GIT and the plasma, then the theoretical ratio R of drug concentration on either
side of the membrane can be given by equations derived by Shore et al:
WEAK ACIDS
WEAK BASE
3/6/2024
20. Drugs pKa pH/site of absorption
Very weak acids
e.g. Pentobarbital
Hexobarbital
>8
Unionized at all pH values; Absorbed along the
entire length of GIT
Moderately weak acids
e.g. aspirin
Ibuprofen 2.5 – 7.5
Unionized in gastric pH & ionized in intestinal pH;
better absorption from stomach
Stronger acids
E.g. disodium Cromogylate < 2.0
Ionized at all pH values; Poorly absorbed from GIT
Very weak bases
e.g. Theophylline
Caffeine
< 5.0
Unionized at all pH values; Absorbed along entire
GIT
Moderately weak bases
e.g. codeine 5 – 11
Ionized at gastric pH, unionized at intestinal pH;
better absorption from intestine.
Stronger bases
e.g. Guanethidine > 11
Ionized at all pH values; Poorly absorbed from GIT
3/6/2024
22. Total aqueous solubility, ST, of an IONISABLE DRUG is an important factor in the passive
absorption of drugs.
It is defined as the SUM OF CONCENTRATION OF IONISED DRUG IN SOLUTION AND
CONCENTRATION OF UNIONISED DRUG IN SOLUTION.
The solubility of unionised form of the drug is known as the intrinsic solubility of the
drug. If Sa is the intrinsic solubility of weakly acidic drugs and Sb that of weakly basic
drugs,
TOTAL AQUEOUS SOLUBILITY
ACIDS
BASE
3/6/2024
24. Lipophilicity and Drug Absorption
The degree of ionisation at a particular pH and that only the unionised drug, if
sufficiently lipid soluble, is absorbed into the systemic circulation.
if the drug exists in the unionised form, it will be poorly absorbed if it has poor lipid
solubility (or low Ko/w)
Systemic Circulation
GASTROINTESTINAL
TRACT
CELL
MEMBRANCE
3/6/2024
26. Limitations of the pH-partition hypothesis
1. Presence of virtual membrane pH
2. Absorption of ionised drug
3. Influence of GI surface area and residence time of drug
4. Presence of aqueous unstirred diffusion layer
PRESENCE OF VIRTUAL MEMBRANE pH
3/6/2024
27. PRESENCE OF AQUEOUS UNSTIRRED DIFFUSION LAYER
Aqueous Bulk Fluid of the GIT
Aqueous Unstirred Layer
Lipoidal Layer (Cell Membrane)
Blood
Limitations of the pH-partition hypothesis
3/6/2024
28. ABSORPTION OF IONISED DRUG
INFLUENCE OF GI SURFACE AREA AND RESIDENCE TIME OF DRUG
Limitations of the pH-partition hypothesis
3/6/2024
29. DRUG PERMEABILITY AND ABSORPTION
M = amount of drug absorbed
Peff = effective membrane permeability
A = surface area available for absorption
Capp = Apparent Luminal Drug Concentration
tres = residence time of Drug in GI lumen
The three major drug characteristics that determine the passive transport or
permeability of drugs across intestinal epithelium are
Lipophilicity of drug expressed as log P.
Polarity of drug which is measured by the number of H-bond acceptors and number of
H-bond donors on the drug molecule.
Molecular size.
3/6/2024
30. Rule of Five by LIPINSKI ET AL which is written as –
Molecular weight of drug ≤ 500
Lipophilicity of drug, log P ≤ 5
Number of H-bond acceptors ≤ 10
Number of H-bond donors ≤ 5
For a given drug, if any two of these values is greater than that specified above,
then oral absorption may be significant problem.
DRUG PERMEABILITY AND ABSORPTION
3/6/2024
31. Dosage form, dosage form related, pharmaceutical excipient
(Pharmaco Technical Factor)
1. Disintegration time (tablets/capsules)
2. Dissolution time
3. Manufacturing variables
4. Pharmaceutical ingredients (excipients/adjuvants)
5. Nature and type of dosage form
6. Product age and storage conditions
PHARMACEUTICAL FACTOR
3/6/2024
32. 1. Disintegration Time:
Coated tablets especially sugar coated have longer DT. DT is directly
proportional to the amount of binder present and the
compressional force of the tablet.
2. Manufacturing /Process variables:
Manufacturing process that affects the DT are :
a. Method of granulation.
b. Compressional force.
3/6/2024
33. 3. Pharmaceutical ingredients:
Excipients used to ensure the acceptability, physicochemical stability during the
shelf life, uniformity of composition and dosage, and optimum bioavailability
and functionality of the drug product.
a. Vehicles b. Diluents
c. Binder and granulating agents d. Disintegrants
e. Lubricants f. Coating
g. Suspending Agents h. Surfactants
4. Nature and type of Dosage form:
As a general rule, the bioavailability of a drug from various dosage form
decreases in the following order:
Solution > Emulsion > Suspension > Capsules > Tablets > Coated tablets > Enteric
Coated tablets > Sustained Release products.
3/6/2024
34. PATIENT RELATED FACTORS AFFECTING DRUG ABSORPTION
Gastrointestinal Tract
The gastrointestinal tract is a muscular tube approximately 450 m in length with
varying diameters.
It stretches from the mouth to the anus and consists of four main anatomical areas:
the oesophagus, the stomach, the small intestine and the large intestine or colon.
The majority of the gastrointestinal epithelium is covered by a layer of mucous. This
is a viscoelastic translucent aqueous gel that is secreted through out the GIT, acting
as a protective layer and a mechanical barrier.
3/6/2024
36. Gastrointestinal Tract:
Organs pH Membrane Blood Flow
L/min
Surface
Area
Sq M
Transit Time
hrs
By-pass
liver
Buccal 6.8 to
7.00
thin Good, fast
absorption with
low dose
small Short unless
controlled
yes
Oesophagus 5-6 Very thick
no
absorption
- small short, typically
a few seconds,
except for
some coated
tablets
-
Stomach 1.0 to
3.00
normal 0.15 small
(0.1 to 0.2 )
30 min (liquid)
- 120 min
(solid food)
Apprx. 1 to 5
hrs
no
Duodenum 5 - 7 normal Good Very large very short, no
3/6/2024
37. Organs pH Membrane Blood
Flow
L/min
Surface
Area
Sq M
Transit Time
hrs
By-pass
liver
Small Intestine 5– 7.5 normal Good
1.0
Very large
200
3-6 no
Large intestine 6.0 – 8.0 - Good
0.02
Not very
large
0.15
6-12 Lower
colon,
rectum yes
Rectum 6.0 – 8.0 - -- 0.02 6-12 Lower
colon,
rectum yes
3/6/2024
38. The environment within the lumen:
Gastrointestinal pH
- As we observed from the previous tables, the pH of fluids varies along the
length of the GIT.
- The gastrointestinal pH may influence the absorption of drugs in a variety of
ways:
A- It may affect the chemical stability of the drug in the lumen e.g. penicillin G,
erythromycin
B- affect the drug dissolution or absorption e.g. weak electrolyte drug
Luminal enzymes
- The primary enzyme found in gastric juice is pepsin. Lipases, amylases and
proteases are secreted from the pancreas into the small intestine.
- Pepsins and proteases are responsible for the digestion of protein and
peptide drugs in the lumen.
Characteristics of GI physiology and Drug Absorption (cont.):
3/6/2024
39. - The lipases may affect the release of drugs from fat / oil –
containing dosage forms.
- Bacteria which are localized within the colonic region of the
GIT secrete enzymes which are capable of a range of
reactions.
- e.g. Sulphasalazine which is a prodrug used to target the
colon.
Sulphasalazine active drug
(5-aminosalycylic acid)
treat inflammatory bowel disease
I. Characteristics of GI physiology and Drug Absorption
(cont.):
Bacterial enzymes
3/6/2024
40. PATIENT RELATED FACTORS
I. Age
II. Gastric Emptying
Factors influence gastric emptying:
1. Volume of Meal
2. Composition of Meal
3. Physical State and Viscosity of Meal
4. Temperature of Meal
5. Gastrointestinal pH
6. Electrolyte and Osmotic Pressure
7. Body Posture
8. Disease State
9. Exercise, emotional state
10. Drugs
III Intestinal Transit
3/6/2024
41. The time a dosage form takes to traverse the stomach is usually
termed: the gastric residence time, gastric emptying time or gastric
emptying rate.
3/6/2024
42. II Gastric emptying and motility:
Factors Affecting Gastric Emptying
3/6/2024
43. II Gastric emptying and motility:
Factors Affecting Gastric Emptying
Viscosity Rate of emptying is greater for less viscous
solutions
Emotional states - Stressful emotional states increase
stomach contraction and emptying rate
- Depression reduces stomach contraction
and emptying
Disease states -Rate of emptying is reduced in:
Some diabetic patients, hypothyrodism
-Rate of emptying is increased in:
hyperthyrodism
Excercise Reduce emptying rate
3/6/2024
44. Presystemic metabolism:
Definition:The metabolism of orally administered drugs by
gastrointestinal and hepatic enzymes, resulting in a significant
reduction of the amount of unmetabolized drug reaching the
systemic circulation.
Gut wall metabolism
- This effect is known as first-pass metabolism by the intestine.
- Cytochrome P450 enzyme, CYP3A, that is present in the liver
and responsible for the hepatic metabolism of many drugs, is
present in the intestinal mucosa and that intestinal metabolism
may be important for substrates of this enzyme e.g.
cyclosporin.
3/6/2024
45. Presystemic metabolism
Hepatic metabolism
- After a drug is swallowed, it is absorbed by the digestive system
and enters the hepatic portal system. It is carried through the
portal vein into the liver before it reaches the rest of the body.
- The liver metabolizes many drugs (e.g. propranolol), sometimes
to such an extent that only a small amount of active drug
emerges from the liver to the rest of the circulatory system.
- This first pass through the liver thus greatly reduces the
bioavailability of the drug.
3/6/2024