Session Objectives
What isVD?
List factors affecting drug distribution.
Identify physiology barriers of drug distribution
4.
Drug Distribution
Involves thetransport of drug molecules within
the body
Drug distribution means the reversible transfer of
drug from one location to another within the
body.
Following absorption or systemic administration
into the bloodstream, a drug distributes into
interstitial and intracellular fluids.
Mechanisms of drugdistribution
Passive diffusion Drug molecules move from an area
of high concentration to an area of low concentration
Most drugs
Hydrostatic pressure -The pressure gradient between
the arterial end of the capillaries entering the tissue
and the venous capillaries leaving the tissue
Hydrostatic pressure is responsible for penetration
of water-soluble drugs into spaces between
endothelial cells and possibly into lymph
Rapid and efficient for Water soluble drugs
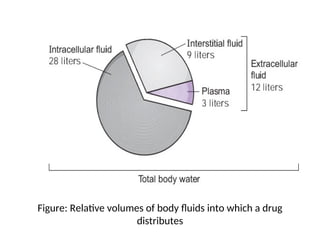
The body isdivided into two spaces, a central and a tissue
compartment.
Central volume (Vc) - a hypothetical volume into which a
drug initially distributes upon administration
- blood in vessels and highly perfused tissues
Peripheral volume (Vt) - the sum of all tissue spaces
outside the central compartment
9.
Together, Vc andVt create the apparent volume of
distribution (Vd).
Apparent Vd - the volume of fluid that would be
required to account for all drug in the body
Distribution volumes are important for estimating:
Amount of drug in the body
Peak serum levels
Clearance
10.
Factors Affecting DrugDistribution
Rate of distribution
Membrane permeability
Blood perfusion
Extent of Distribution
Lipid Solubility
Plasma protein binding
Tissue protein binding
11.
Lipid Solubility
Lipid solubilitywill affect the ability of the drug
to bind to plasma proteins and to cross lipid
membrane barriers.
Large depots of drug in fat may necessitate a
longer period of time for drug to be removed
from the body.
The distribution of lipophilic drugs will be
different in thin versus obese patients.
12.
Membrane permeability
Lipid solubledrugs pass through very rapidly.
Water soluble compounds penetrate more
slowly at a rate more dependent on their size.
Low molecular weight drugs pass through by
simple diffusion.
13.
Permeability is greatlyincreased in the renal and
hepatic capillaries
Brain capillaries seem to have impermeable walls
restricting the transfer of molecules from blood to
brain tissue.
Lipid soluble compounds can be readily transferred but
the transfer of polar substances is severely restricted.
This is the basis of the "blood-brain" barrier.
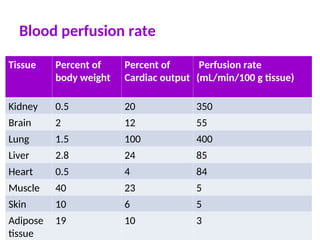
Rapidly perfused tissuesrespond quickly
Bain
Liver
Kidney
Less rapidly perfused tissues respond to drug more slowly
Muscle
Skin
Poorly perfused tissues respond very slowly to drug
Fat
Organs with high blood flow will experience larger initial
effects
16.
Plasma protein binding
Extensiveplasma protein binding will cause more
drug to stay in the central blood compartment.
Therefore drugs which bind strongly to plasma
protein tend to have lower volumes of distribution
The extent of this binding will influence the drug’s
distribution and rate of elimination
only the unbound drug can diffuse through the wall,
produce its systemic effects, be metabolized, and be
excreted.
17.
Most drugs forma complex with proteins
D + P ↔DP (reversible binding)
Bound drug is in equilibrium with free drug.
Free drug is active and bound drug is inactive.
More free drug when binding sites are saturated.
Competition between drugs for binding sites.
Protein binding allows a part of a drug dose to be
stored and released as needed
18.
Protein binding ofdrugs
Some drugs are highly bound (> 90%) to plasma
proteins.
Slight changes in the binding of highly bound drugs
can result in significant changes in clinical response
or cause a toxic response.
Example: warfarin and phenytoin
Acidic drugs commonly bind to albumin, while basic
drugs often bind to α1-acid glycoproteins and
lipoproteins.
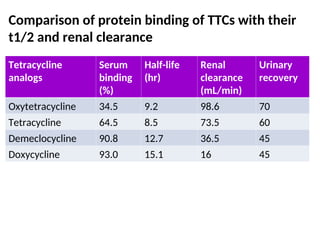
Comparison of proteinbinding of TTCs with their
t1/2 and renal clearance
Tetracycline
analogs
Serum
binding
(%)
Half-life
(hr)
Renal
clearance
(mL/min)
Urinary
recovery
Oxytetracycline 34.5 9.2 98.6 70
Tetracycline 64.5 8.5 73.5 60
Demeclocycline 90.8 12.7 36.5 45
Doxycycline 93.0 15.1 16 45
21.
Determinants of proteinbinding
The drug
Physicochemical property of the drug
Total concentration of the drug in the body
The protein
Quantity of the protein available for drug protein
interaction
Quality or physicochemical nature of the protein
synthesized
Affinity b/n drug and protein
22.
Drug interaction
Competitionfor the drug by other substances at a protein-
binding site
Alteration of a protein by a substance that modifies the
affinity of the drug for the protein
aspirin acetylates lysine residue of albumin
5. The pathophysiologic condition of the patient
Example: uremic and hepatic patients
23.
Protein Binding Interaction
Onedrug may displace another from the same
binding site
Free drug concentration is usually the important
factor
Increase activity
increase elimination
Eg. Phenylbutazone displaces tolbutamide
24.
Disease state decreaseplasma protein
concentration
Liver disease-decrease protein synthesis
Trauma, surgery- increase protein catabolism
Burns- increase distribution of albumin into
extracellular space
Renal disease- increase excessive elimination of
protein
24
25.
Tissue localization ofdrugs
Drugs will not always be uniformly distributed to
and retained by body tissues.
The concentrations of some drugs will be either
higher or lower in particular tissues than could be
predicted on the basis of simple distribution
assumptions.
26.
Kidney:
The kidney containsa protein, metallothionein, that has a
high affinity for metals.
This protein is responsible for the renal accumulation of
cadmium, lead, and mercury
Eye.
Several drugs have an affinity for the retinal pigment
melanin and thus may accumulate in the eye.
Example: Chlorpromazine, Chloroquine .
Lung.
The lung receives the entire cardiac out-put
Most compounds that accumulate in the lung are basic
amines
Examples: antihistamines, imipramine, amphetamine,
methadone, and chlorpromazine
27.
Fat
Drugs with extremelyhigh lipid–water partition
coefficients have a tendency to accumulate in body fat
like DDT
But into body fat occurs slowly
Drug accumulation in body fat may result either in
decreased therapeutic activity owing to the drug’s
removal from the circulation or
in prolonged activity when only low levels of the
drug are needed to produce therapeutic effects
Bone:
Although bone is a relatively inert tissue, it can accumulate
such substances as tetracyclines, lead, strontium, and the
antitumor agent cisplatin.
28.
Physiologic barriers ofdistribution
Most capillaries have pores between the endothelial cells
lining the capillaries
In some capillary beds, however, the endothelial cells are
closely connected by “tight junctions”, and such
capillaries do not have pores between the endothelial
Only lipophilic drugs rapidly diffuse across capillary
beds with tight junctions, whereas hydrophilic drugs are
mostly excluded.
29.
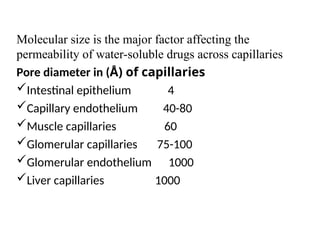
Molecular size isthe major factor affecting the
permeability of water-soluble drugs across capillaries
Pore diameter in (Å) of capillaries
Intestinal epithelium 4
Capillary endothelium 40-80
Muscle capillaries 60
Glomerular capillaries 75-100
Glomerular endothelium 1000
Liver capillaries 1000
30.
The “blood-brain barrier(BBB)”
Capillaries in brain have:
tight junctions per capillary gelial cells
p-glycoprotein: back to the systemic
circulation
All contribute to BBB
The BBB restricts the movement of hydrophilic drugs
into brain; however, the BBB is “broken” by ischemia
and inflammation
31.
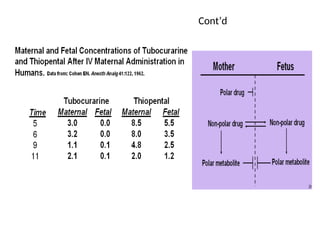
Passage of drugsacross the placenta
Capillary walls separating fetal blood from maternal
blood are continuous
The placenta is not an effective barrier to most drugs
Many drugs can be found in fetus shortly after the
administration to mother
fetus can be pharmaceutically treated through mother’s
body
risk of the undesirable effects is high
32.
In general, substancesthat are lipid soluble
cross the placenta with relative ease in
accordance with their:
lipid–water partition coefficient and
degree of ionization.
Highly polar or ionized drugs do not cross the
placenta readily.
However, most drugs used in labor and
delivery are not highly ionized and will cross.
33.
They are generallyweak bases with pKa values of
about 8 and tend to be more ionized in the fetal
bloodstream, since the pH of fetal blood is around
7.3 as compared with the maternal blood pH of
7.44.
Differences in maternal and fetal blood pH can
give rise to unequal concentrations of ionizable
drugs in the mother and the fetus
Factors that mayinfluence placental transfer
Factor Effect
Placental blood flow Increased delivery of the drug to the
placenta
Molecular size of the drug Decrease in delivery as size increases
Impermeable: MW> 1000
Permeable: MW<600
Lipid solubility of drug Increase transfer as lipid solubility
increases
pKa of the drug Ion trapping on either side
37
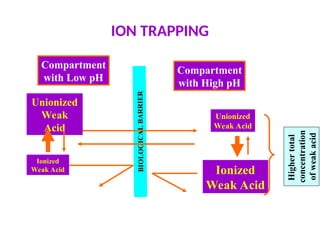
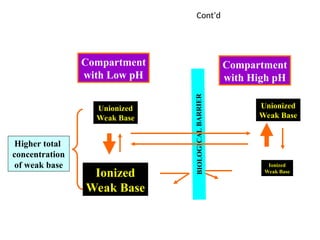
cont’d
Ion trappingcan be used to distribute drugs into the urinary
compartment to increase the urinary excretion of poisons.
-Alkalinization of the urine with systemic administration of
sodium bicarbonate is useful for the treatment of overdoses of
aspirin and phenobarbital.
-Acidification of the urine with systemic administration of
ammonium chloride is useful for the treatment of amphetamine
overdoses.
DRUG ELIMINATION
Drug elimination
refers to the irreversible removal of drug from the
body by all routes of elimination.
Drugs are removed from the body by various
elimination processes.
Both metabolism and excretion can be viewed as
processes responsible for elimination of drug
(parent and metabolite) from the body.
42
43.
Kinetics of elimination
Kineticsof elimination
F
First-order
irst-order elimination or kinetics
elimination or kinetics
For most drugs, the rate of elimination from the body is
proportional to the amount of drug present in the body
(AB).
This type of elimination kinetics is called first-order
elimination or kinetics.
The elimination rate constant (kel) is used to denote how
quickly drug serum concentrations decline in a patient.
43
44.
With first-orderelimination, the amount of drug eliminated
over a certain time period increases as the amount of drug
in the body increases; likewise,
the amount of drug eliminated per unit of time decreases as
the amount of drug in the body decreases.
If the amount of drug in the body is known, the elimination
rate for the drug can be computed by taking the product of
the elimination rate constant and the amount of drug in the
body (AB).
44
45.
The rate ofelimination of the drug that follows first
order elimination can be described as:
Elimination rate = dA/dt = - k A ,
where k is the first-order rate constant.
With first order elimination,
Elimination rate is dependent on the concentration
of A present in the body.
45
46.
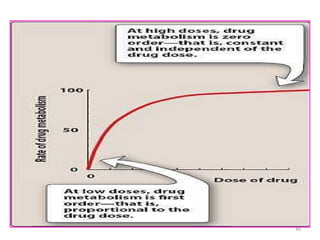
Zero-order elimination
Zero-order elimination
If large amount of drug is administered, then order of
elimination process of the drugs will change from a first-order
process to a zero-order process.
With zero-order elimination,
The amount of drug eliminated does not change with AB
The fraction removed varies
The rate of elimination of the drug that follows zero order
elimination can be described as:
elimination rate = dA/dt = - k*, where k* is the zero-order
rate constant
46
47.
Zero-order elimination occurswhen the body's
ability to eliminate a drug has reached its
maximum capability (i.e., all transporters are being
used).
As the dose and drug concentration increase, the
amount of drug eliminated per hour does not
increase, and the fraction of drug removed
declines.
A few substances are eliminated by zero-order
elimination kinetics, because their elimination
process is saturated.
47
48.
Because in asaturated process the elimination rate
is no longer proportional to the drug concentration
zero-order kinetics are also called “non-linear
kinetics in clinical pharmacology.
48
Metabolism
Drug metabolism changesthe chemical structure of a
drug to produce a drug metabolite, which is frequently
but not universally less pharmacologically active.
Metabolism also renders the drug compound more
water soluble and therefore more easily excreted.
The liver
liver is the major site for drug metabolism, but
specific drugs may undergo biotransformation in other
tissues, such as the kidney and the intestines
kidney and the intestines.
50
51.
Drug metabolism reactionsare carried out by enzyme
systems which are important to protect the body from
exogenous chemicals.
The enzyme systems for this purpose for the most part
can be grouped into two categories:
Phase I oxidative or reductive enzymes
Phase II conjugative enzymes.
51
52.
Phase I :-oxidative and reductive enzymes:
Phase I enzymes act by causing the drug molecule to
undergo oxidation or more rarely, reduction.
Reaction Examples
Aliphatic and aromatic
hydroxylation
Ibuprofen, flurbiprofen
N-demethylation Morphine
O-demethylation Codeine
Epoxidation Carbamazepine
N-Oxidation Morphine
S-Oxidation Sulindac
Deamination Amphetamine 52
53.
Cytochrome P450 Enzymes
Thecytochrome P450 (CYP450) enzyme super
family is the primary phase I enzyme system involved
in the oxidative metabolism of drugs and other
chemicals.
These enzymes also are responsible for all or part of
the metabolism and synthesis of a number of
endogenous compounds, such as steroid hormones
and prostaglandins.
To date, 12 unique isoforms have been identified as
playing a role in human drug metabolism.
53
54.
CYP
Isoform
Examples of Substrates
CYP1A1Essentially same as CYP1A2
CYP1A2 Polycyclic aromatic hydrocarbons, caffeine, theophylline
CYP2A6 Nicotine, 5-fluorouracil, coumarin
CYP2B6 Bupropion, cyclophosphamide, propofol
CYP2C8 Paclitaxel
CYP2C9 Phenytoin, warfarin, nonsteroidal antiinflammatory drugs
CYP2C19 Omeprazole
CYP2D6 Tricyclic antidepressants, codeine, dextromethorphan, some b-
blockers, some antipsychotics, some antiarrhythmics
CYP2E1 Acetaminophen, chlorzoxazone
CYP3A4 Midazolam, triazolam, cyclosporine, erythromycin, HIV protease
inhibitors, calcium channel blockers
CYP3A5 Essentially same as CYP3A4
CYP3A7 Unclear but may be similar to CYP3A4
54
55.
• More thanone CYP isoform may be involved in
the metabolism of a particular drug.
– Verapamil is primarily metabolized by CYP3A4, but
CYPs 2C9, 2C8 and 2D6 participate to some degree,
particularly in the 20
metabolism of the verapamil
metabolites.
55
56.
Substrate Specificity ofthe CYP Enzymes
CYP3A4
the most predominant CYP isoform, both in terms of
the amount of enzyme in the liver and the variety of
drugs
Account for more than 50% of all CYP-mediated drug
oxidation reactions,
involved in the greatest number of drug–drug
interactions.
The active site of CYP3A4 is thought to be large
relative to other isoforms, as evidenced by its ability to
accept substrates up to a molecular weight of 1200
(e.g., cyclosporine).
56
57.
CYP3A5:
Amino acid sequenceis similar to that of CYP3A4
Same substrate specificity characteristics as CYP3A4.
Not present in all individuals.
CYP3A7:
Appears to be expressed only in the fetus and rapidly
disappears following birth, to be replaced by CYP3A4
and CYP3A5.
CYP2D6:
The second most common CYP isoform
Accounts for 30% of the CYP-mediated oxidation
reactions
57
58.
CYP2C9
About 10% ofthe CYP-mediated drug
oxidation
Metabolizes several clinically important
drugs with narrow therapeutic indices.
-Warfarin, phenytoin
58
59.
Enzyme Inhibition
Enzyme inhibitionis the most frequently observed
result of CYP modulation and is the primary
mechanism for drug–drug pharmacokinetic
interactions.
Two types:
Simple competitive inhibition
Mechanism-based inactivation (or suicide inactivation)
59
60.
Simple competitive inhibition
Twodrugs are competing for the same active site
The drug with the highest affinity for the site wins
out
Addition of a second drug with greater affinity for the
enzyme inhibits metabolism of the primary drug
Results in an elevated primary drug blood or tissue
concentration
Example: ketoconazole and triazolam compete for
binding to the CYP3A4
17 Xs in conc. of triazolam
60
61.
Mechanism-based inactivation
suicide inactivation
theeffector compound (the inhibitor) is itself
metabolized by the enzyme to form a reactive species
that binds irreversibly to the enzyme and prevents any
further metabolism by the enzyme.
61
62.
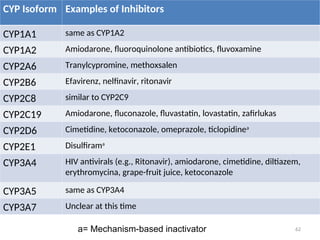
CYP Isoform Examplesof Inhibitors
CYP1A1 same as CYP1A2
CYP1A2 Amiodarone, fluoroquinolone antibiotics, fluvoxamine
CYP2A6 Tranylcypromine, methoxsalen
CYP2B6 Efavirenz, nelfinavir, ritonavir
CYP2C8 similar to CYP2C9
CYP2C19 Amiodarone, fluconazole, fluvastatin, lovastatin, zafirlukas
CYP2D6 Cimetidine, ketoconazole, omeprazole, ticlopidinea
CYP2E1 Disulfirama
CYP3A4 HIV antivirals (e.g., Ritonavir), amiodarone, cimetidine, diltiazem,
erythromycina, grape-fruit juice, ketoconazole
CYP3A5 same as CYP3A4
CYP3A7 Unclear at this time
62
a= Mechanism-based inactivator
63.
Enzyme Induction
Induction ofdrug-metabolizing activity can be due
to:
Synthesis of new enzyme protein or
A decrease in the proteolytic degradation of the
enzyme.
Net result of enzyme induction is the increased
turnover (metabolism) of substrate.
Results in therapeutic failure
63
64.
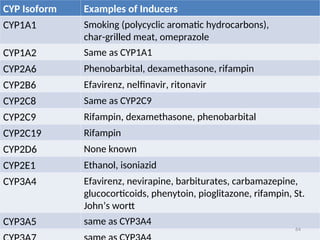
CYP Isoform Examplesof Inducers
CYP1A1 Smoking (polycyclic aromatic hydrocarbons),
char-grilled meat, omeprazole
CYP1A2 Same as CYP1A1
CYP2A6 Phenobarbital, dexamethasone, rifampin
CYP2B6 Efavirenz, nelfinavir, ritonavir
CYP2C8 Same as CYP2C9
CYP2C9 Rifampin, dexamethasone, phenobarbital
CYP2C19 Rifampin
CYP2D6 None known
CYP2E1 Ethanol, isoniazid
CYP3A4 Efavirenz, nevirapine, barbiturates, carbamazepine,
glucocorticoids, phenytoin, pioglitazone, rifampin, St.
John’s wortt
CYP3A5 same as CYP3A4
64
65.
Phase II-Conjugative Enzymes:
PhaseII conjugative enzymes metabolize drugs by
attaching (conjugating) a more polar molecule to
the original drug molecule to
increase water solubility, thereby permitting
more rapid drug excretion.
65
66.
Glucuronosyl Transferases
UGTs conjugatethe drug molecule with a
glucuronic acid moiety
Through establishment of an ether, ester, or
amide bond.
The glucuronic acid moiety, being very water
soluble, generally renders the new conjugate
more water soluble and thus more easily
eliminated.
opioids, androgens, estrogens, progestins, and
nonsteroidal antiinflammatory drugs
66
67.
N-Acetyltransferases
the N-acetyltransferase (NAT)enzymes catalyze
to a drug molecule the conjugation of an acetyl
moiety derived from acetyl coenzyme A.
The net result of this conjugation is an increase
in water solubility and increased elimination of
the compound.
67
68.
Sulfotransferases and Methyltransferases
Sulfotransferases(SULTs): metabolism of a number of
drugs, neurotransmitters, and hormones, especially the
steroid hormones.
The co-substrate for these reactions is 3’-
phosphoadenosine 5’-phosphosulfate (PAPS)
Methyltransferases (MTs): methylation of drugs,
hormones, neurotransmitters, proteins, RNA, and DNA.
MTs use S-adenosyl-L-methionine (SAM) as the methyl
donor
68
69.
Pharmacogenetics of DrugMetabolizing Enzymes
Genetic polymorphism of drug-metabolizing enzymes
Acetylation: rapid acetylators and slow acetylators.
Slow acetylators (about 50% of the caucasian population)
are more prone to adverse effects
69
70.
EXCRETION OF DRUGS
Excretion,along with metabolism and tissue
redistribution, is important in determining both the
duration of drug action and the rate of drug
elimination.
Excretion is a process whereby drugs are transferred
from the internal to the external environment,
The principal organs involved in this activity are the
kidneys, lungs, biliary system, and intestines.
70
71.
RENAL EXCRETION
The kidneyis the primary organ of removal for
most drug, especially for those that are water
soluble and not volatile.
The three principal processes that determine the
urinary excretion of a drug are
glomerular filtration,
tubular secretion, and
tubular reabsorption (mostly passive back-diffusion)
71
72.
Glomerular Filtration
The ultrastructureof the glomerular capillary wall
is such that it permits a high degree of fluid
filtration while restricting the passage of
compounds having relatively large molecular
weights.
This selective filtration is important in that it
prevents the filtration of plasma proteins (e.g.,
albumin) that are important for maintaining an
osmotic gradient in the vasculature and thus
plasma volume.
72
73.
Factors influencing theGFR are:
Molecular size, charge, and shape
Protein bound
Inflammation of the glomerular capillaries
73
74.
Reabsorption
Substances diffuse backacross the tubular
membranes and reenter the circulation.
Reabsorption of water that occurs throughout most
portions of the nephron increased concentration
of drug in the luminal fluid the movement of
drugs from the tubular lumen to blood.
Acidification:
reabsorption (or elimination) of weak acids,
such as salicylates
reabsorption (or elimination) of weak bases,
such as amphetamines 74
75.
Active Tubular Secretion
Anumber of drugs can serve as substrates for the two
active secretory systems in the proximal tubule cells.
One secretes organic anions , and the other secretes
organic cations.
One drug substrate can compete for transport with a
simultaneously administered or endogenous similarly
charged compound
Saturation at higher concentration
75
76.
DRUG CLEARANCE
Drug clearanceis a pharmacokinetic term for describing
drug elimination from the body without identifying the
mechanism of the process.
Drug clearance (body clearance, total body clearance , or
Cl T ) considers the entire body as a single drug-
eliminating system from which many unidentified
elimination processes may occur.
Instead of describing the drug elimination rate in terms of
amount of drug removed per time unit (eg, mg/min), drug
clearance is described in terms of volume of fluid clear of
drug per time unit (eg, mL/min).
76
77.
• Mathematically, clearanceis the division of the rate of
elimination and plasma concentration (Cp).
77
)
L
mg
(
C
)
h
mg
(
n
eliminatio
of
Rate
CL
p
B
el A
.
k
elimin
of
Rate
d
p
V
C
A
. el
el k
k
CL
78.
Drugs can becleared from the body by many different
mechanisms, pathways, or organs, including hepatic
biotransformation and renal and biliary excretion.
Renal clearance = rate of elimination by kidney
C
Hepatic clearance = rate of elimination by liver
C
Other organ clearance = rate of elimination by organ
C
CL total = CL renal + CL hepatic + CL pulmonary +CL others
78
Editor's Notes
#13 i) Permeability is greatly increased in the renal capillaries by pores in the membrane of the endothelial cells, and in specialized hepatic capillaries, known as sinusoids which may lack a complete lining. This results in more extension distribution of many drugs out of the capillary bed.
ii) On the other hand brain capillaries seem to have impermeable walls restricting the transfer of molecules from blood to brain tissue.
#27 Drugs with extremely high lipid–water partition coefficients have a tendency to accumulate in body fat. However, since blood flow
to adipose tissue is low (about 3 mL/100 g/minute), distribution into body fat occurs slowly. Drug accumulation in body fat may result either in decreased therapeutic activity owing to the drug’s removal from the circulation or in prolonged activity when only low levels of the drug are needed to produce therapeutic effects. In the latter instance, fat depots provide a slow, sustained release of the active drug. Should body fat be seriously reduced, as during starvation, stored compound
Lung. The lung receives the entire cardiac out-put; therefore, drug distribution into it is very rapid. Most compounds that accumulate in the lung are basic amines (e.g., antihistamines, imipramine, amphetamine, methadone, phentermine, chlorphentermine, and chlorpromazine) with large lipophilic groups and pK values greater than 8.However, some nonbasic amines, such as the herbicide paraquat, also can accumulate in the lung.
#60 Vie: vying: contend: to strive for superiority or compete with somebody or something
#73 inflammation of the glomerular capillaries may increase GFR
#75 The secretory capacity of both the organic anion and organic cation secretory systems can be saturated at high drug concentrations.