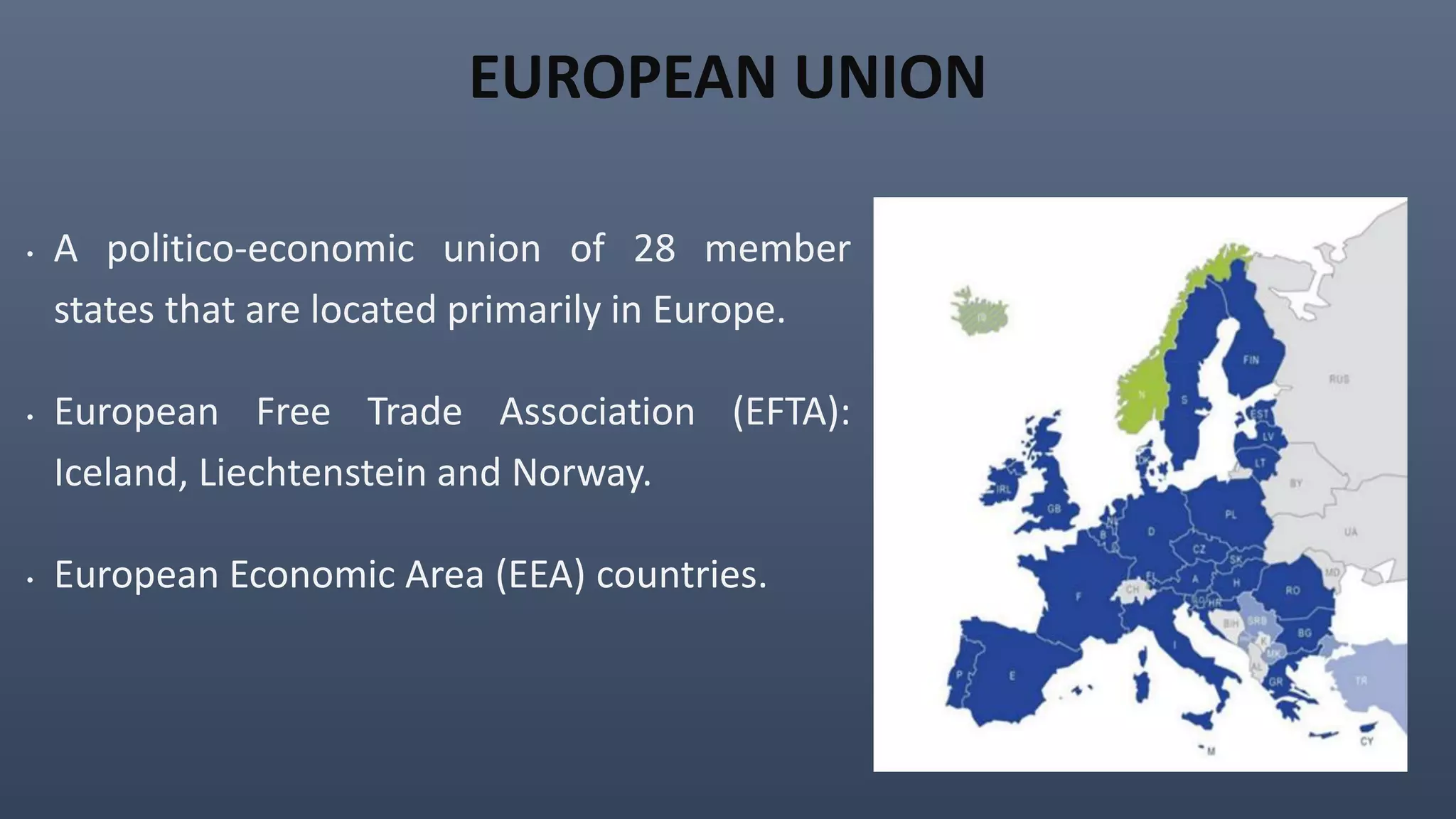
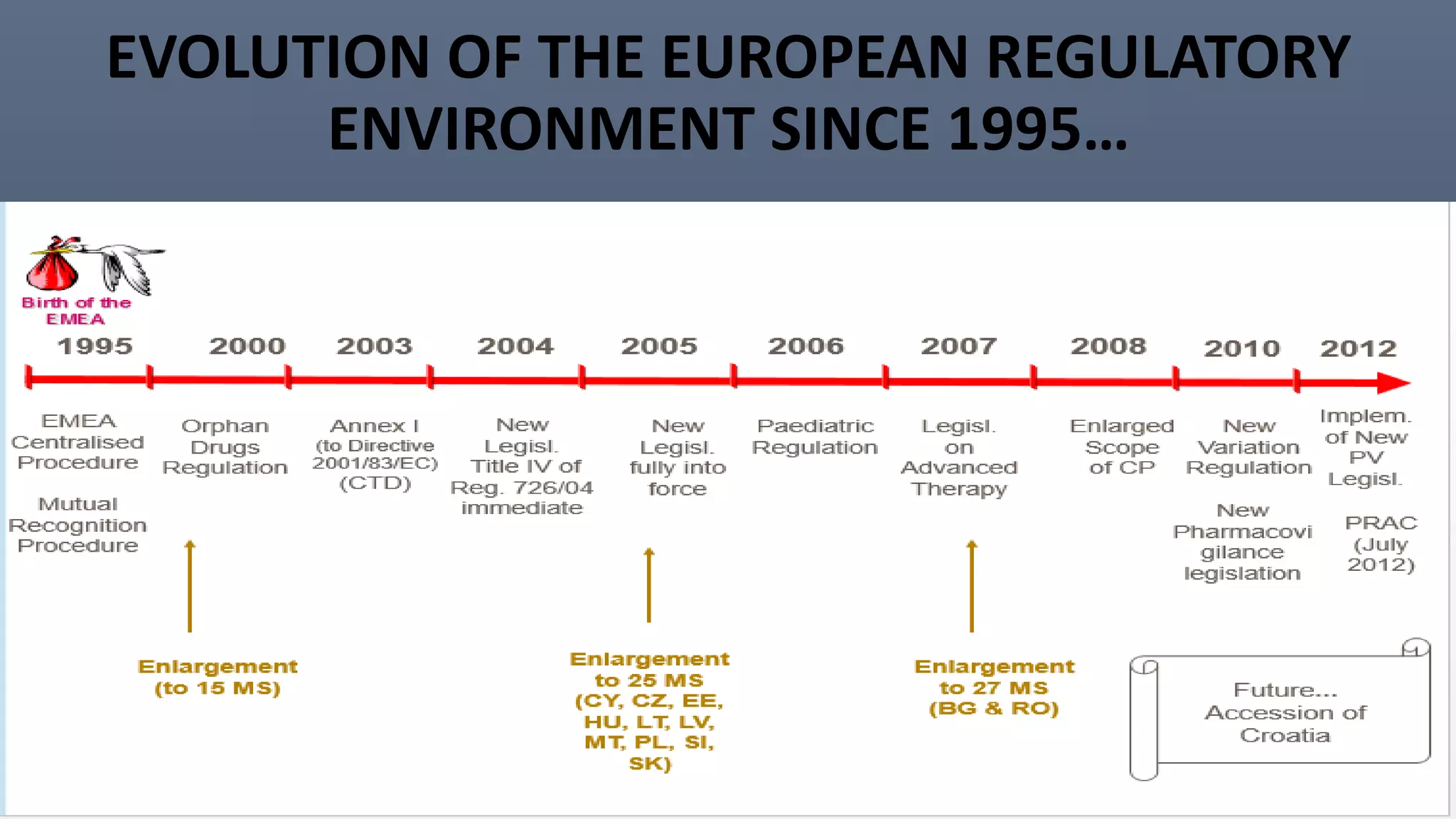
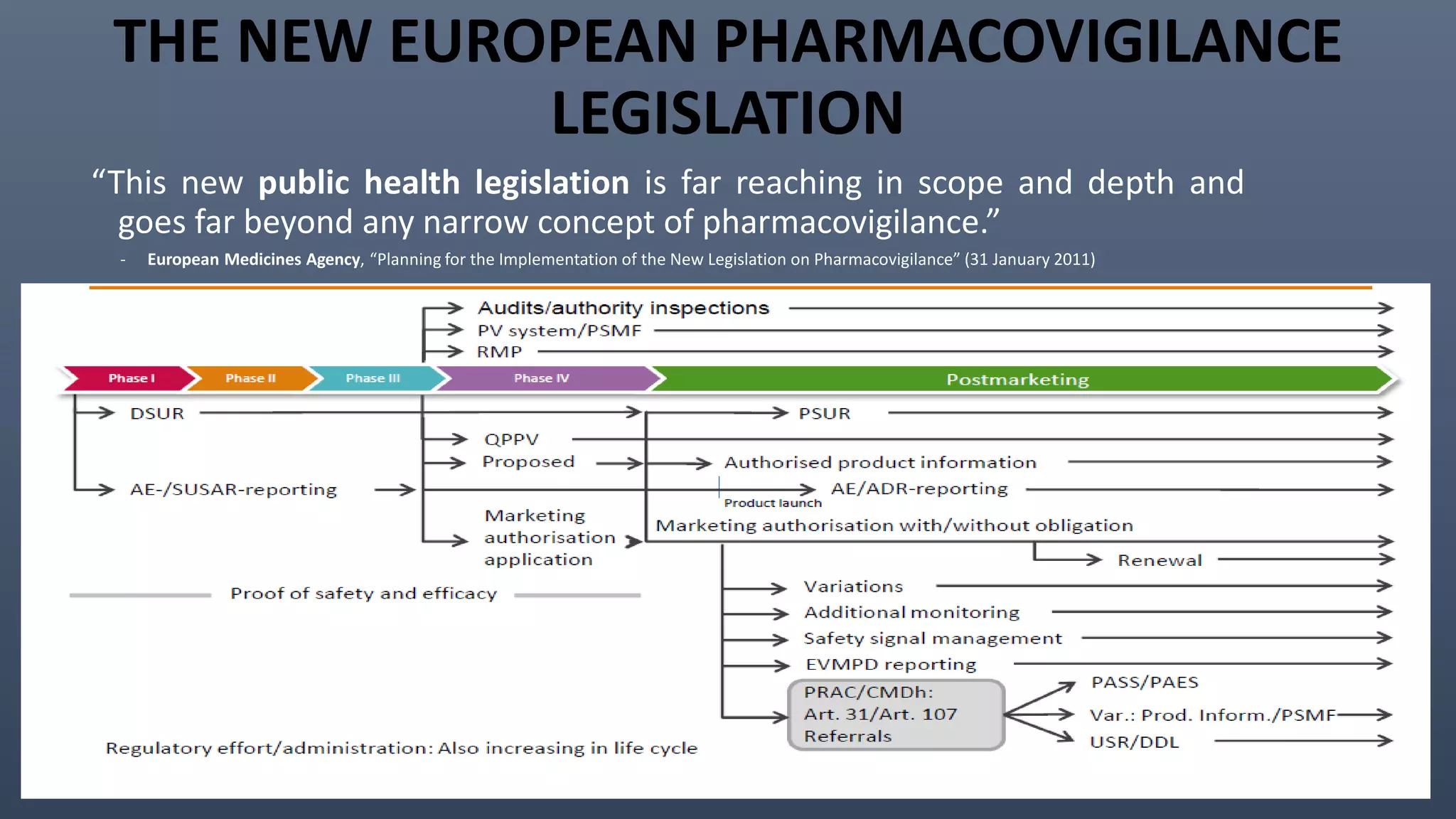
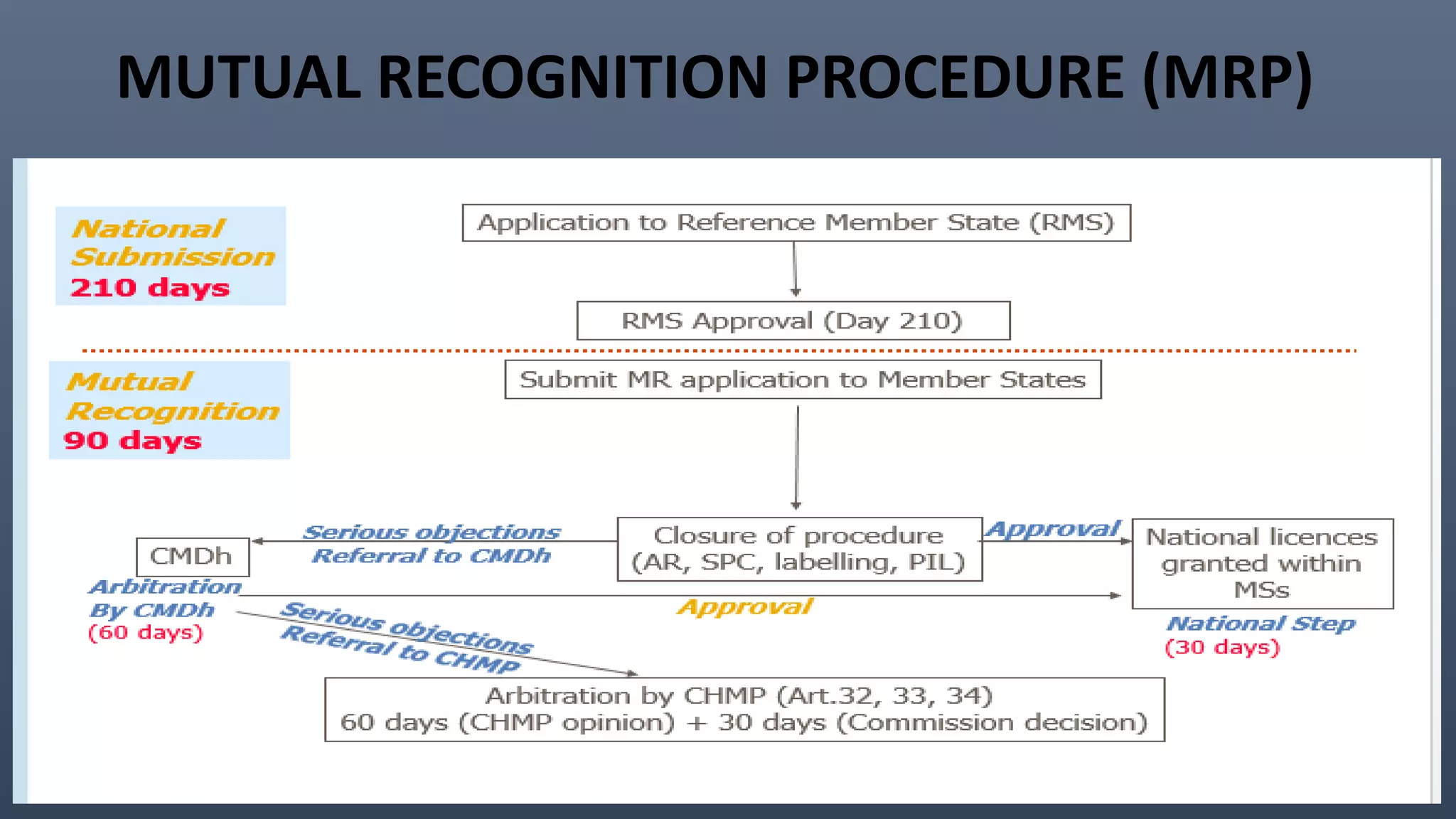
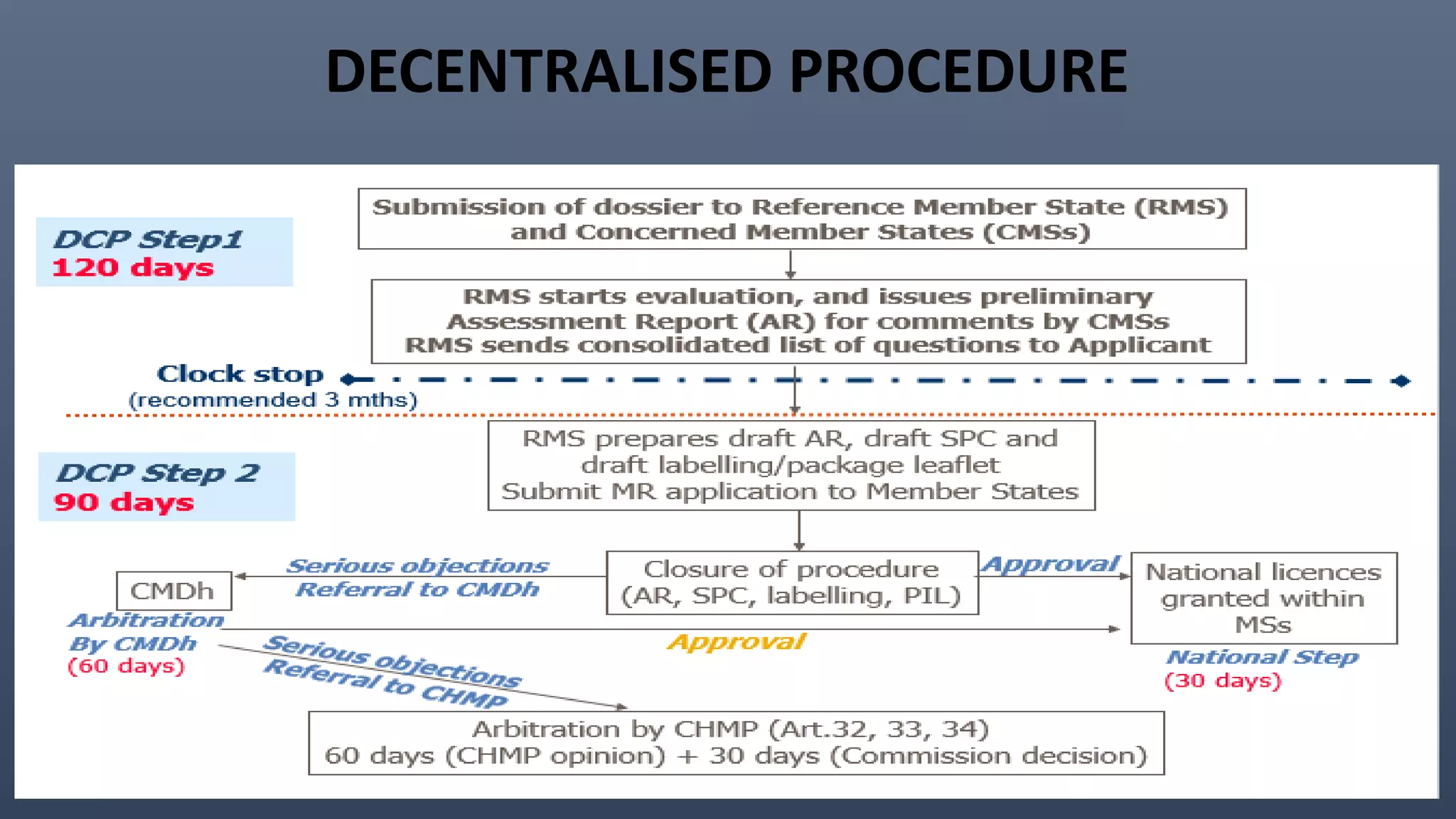
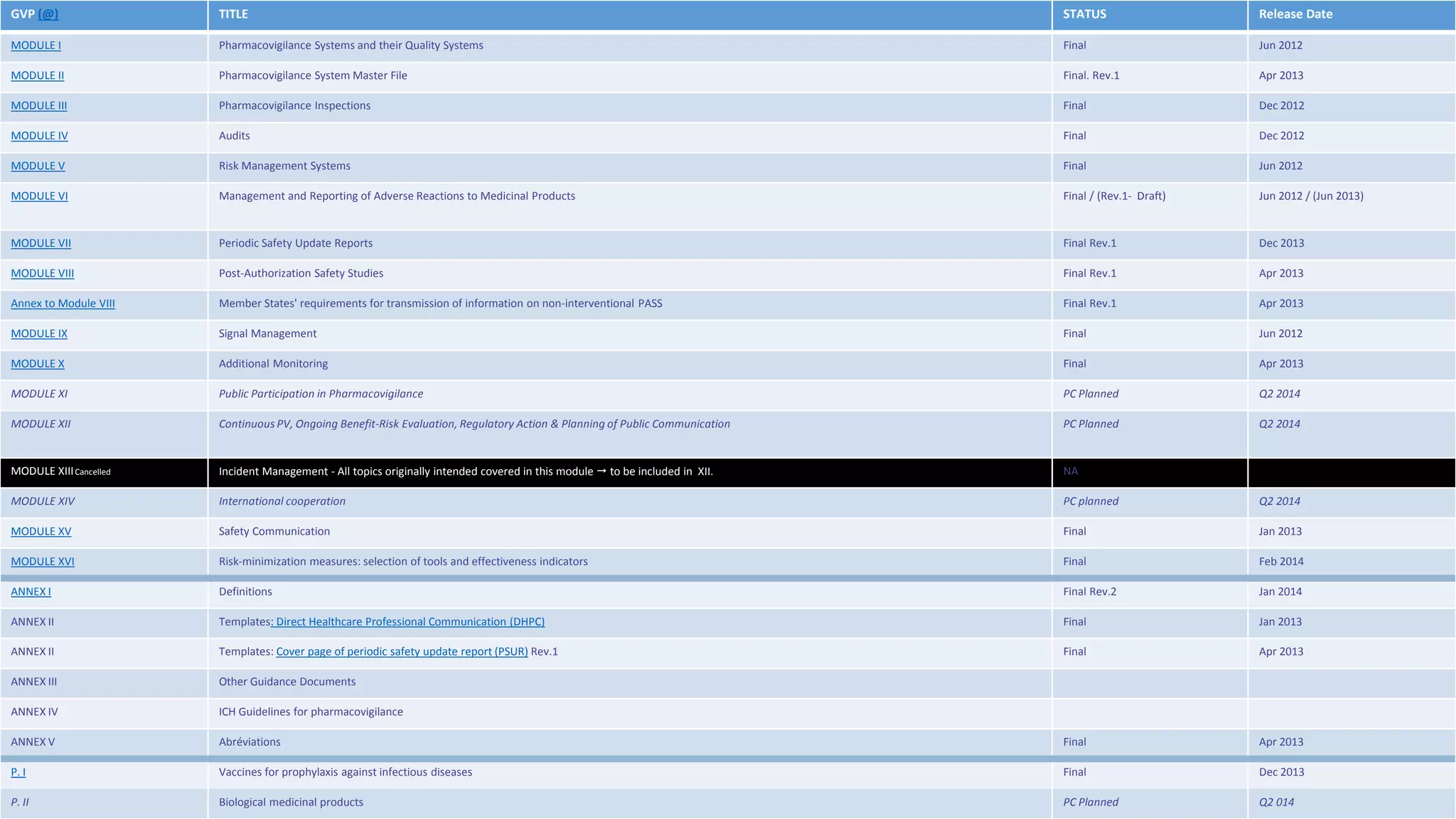
This document discusses the key aspects of pharmacovigilance in the European Union. It provides an overview of the process of monitoring medicine safety, outlines the authorities involved, and describes the evolution of the regulatory environment over time. Major changes introduced by new EU legislation include the Good Pharmacovigilance Practice guidelines, the new Pharmacovigilance Risk Assessment Committee, requirements for a Pharmacovigilance System Master File, changes to periodic safety reports, more stringent rules for reporting adverse events, publishing Risk Management Plan summaries, and the potential for requiring post-approval safety studies. The overall aim is to strengthen the EU pharmacovigilance system and better protect public health.








![2. Pharmacovigilance Risk Assessment Advisory
Committee (PRAC):
• A new EMA committee-meets monthly from September 2012.
• Replaces the Pharmacovigilance Working Party.
• Advises to CHMP and CMDh.
• Members include (appointed by MS and EC) :
• Experts from the EU Member States.
• Representatives from Patient organizations.
• Representatives from Healthcare professionals.
• responsible for assessing all aspects of the risk management of the use of medicinal products
[human use] approved in EEA.
• responsible for the design and evaluation of post-authorisation safety studies and
pharmacovigilance audit.](https://image.slidesharecdn.com/europeanpv-151012191226-lva1-app6891/75/Changes-in-European-PV-post-July-2011-15-2048.jpg)













































![Deschambault[1]](https://cdn.slidesharecdn.com/ss_thumbnails/89882af6-e960-410c-be05-14e4d6adb648-160708012743-thumbnail.jpg?width=640&height=640&fit=bounds)


























![Volume 9 A Guidelines On Pharmacovigilance[1]](https://cdn.slidesharecdn.com/ss_thumbnails/volume9aguidelinesonpharmacovigilance1-12816046179281-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)






























