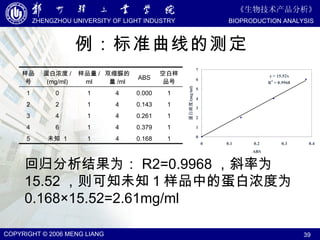
该文档详细介绍了分光光度分析和比色分析的原理、方法及应用。文中涵盖了比尔定律、光谱特性、化学相互作用及噪声对分析结果的影响,并讨论了如何通过标准曲线进行分析。最后,文档还提及了不同类型的分光光度计以及荧光光谱和比色反应的必要条件。
































































![1. 固定时刻动力学分析 优点是使用方便,可使用低值设备,不需要记录仪和类似的设备 缺点是产物的信号不是在多个时间点记录的,分析者不能直接确定随时间具有固定的速率的假设是否正确 0 0.5 1 1.5 2 2.5 3 0 10 20 30 40 时间 /min 吸光度 0 0.5 1 1.5 2 2.5 0 5 10 15 20 25 [ 分析物 ] 吸光度 (10min 后 )](https://image.slidesharecdn.com/bioanalysis03spectrum-2639/85/Bioanalysis-03-Spectrum-66-320.jpg)











![( 1 ) S>>Km 此时米氏方程简化为 v ≈ v max =[E tot ] k cat 则此时可用上式直接进行计算即可](https://image.slidesharecdn.com/bioanalysis03spectrum-2639/85/Bioanalysis-03-Spectrum-78-320.jpg)















![假一级反应 酶催化 当 [S]<<Km 时,米氏方程可简化为一级反应 v =( v max /K m )[S] 非酶催化 当一种反应物浓度远远大于另一种反应浓度时的初速度为假一级反应](https://image.slidesharecdn.com/bioanalysis03spectrum-2639/85/Bioanalysis-03-Spectrum-94-320.jpg)






















![多个酶浓度计算示例 0.112 10.0 0.027 2.5 0.083 7.5 0.012 1.0 0.054 5.0 0 0.0 速度 /[μmol/(ml·min)] 酶浓度 /(μg/ml) 速度 /[μmol/(ml·min)] 酶浓度 /(μg/ml)](https://image.slidesharecdn.com/bioanalysis03spectrum-2639/85/Bioanalysis-03-Spectrum-117-320.jpg)






![不同酶浓度下时间与吸光度数据 标准曲线 /[0.01318AU/(nmol/ml)] 13.35 7.44 4.62 0.00 除以斜率新单位 /[nmol/(ml·min)] 0.17594 0.09803 0.06089 0.00 最小二乘法斜率 /(AU/min) 0.853 0.488 0.312 0 5 0.503 0.29 0.17 0 3 0.317 0.186 0.105 0 2 0.153 0.099 0.076 0 1 0.02 0.01 0.01 0 0 5 2.5 1.5 0 酶浓度 /(μg/ml) 时间 /min](https://image.slidesharecdn.com/bioanalysis03spectrum-2639/85/Bioanalysis-03-Spectrum-124-320.jpg)












![实例:测定兔血清中 IgG 浓度 10.0 2.0 12 0.50 0.05 6 7.5 1.0 11 0.25 0.03 5 5.0 0.50 10 0.10 0.02 4 2.5 0.30 9 0.075 0.01 3 1.0 0.20 8 0.050 0.005 2 0.75 0.10 7 0.025 0 1 [ 兔血清 ]/( μ g/ml) 0.1ml/ 孔 行 C 和 D [ 兔 IgG]/( μ g/ml) 0.1ml/ 孔 行 A 和 B 列 [ 兔血清 ]/( μ g/ml) 0.1ml/ 孔 行 C 和 D [ 兔 IgG]/( μ g/ml) 0.1ml/ 孔 行 A 和 B 列](https://image.slidesharecdn.com/bioanalysis03spectrum-2639/85/Bioanalysis-03-Spectrum-137-320.jpg)




















