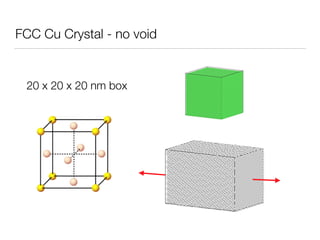
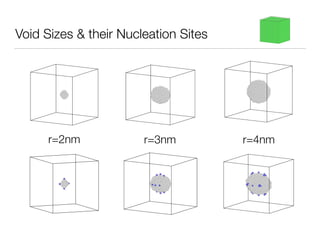
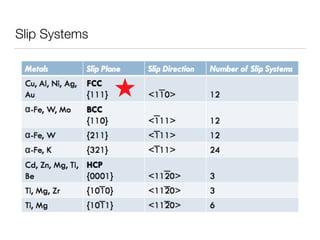
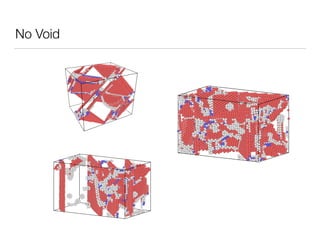
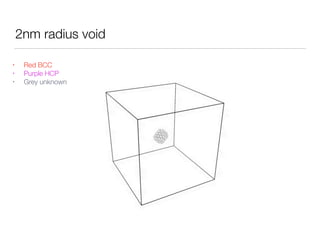
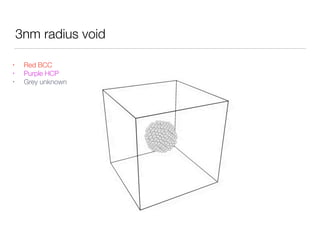
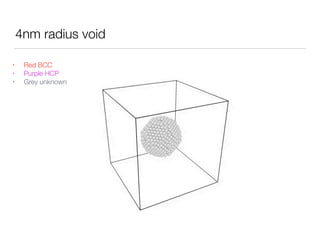
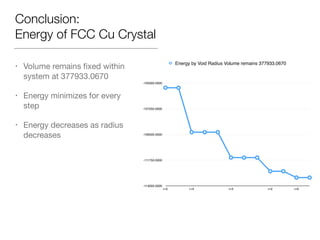
This document summarizes a molecular dynamics simulation of voids in an FCC copper crystal under uniaxial tensile deformation. It describes using LAMMPS to simulate copper crystals with voids of varying radii (2nm, 3nm, 4nm) and analyze the effect on energy levels and dislocation formation. The simulation found that energy decreases as void radius decreases, with the lowest energy structure having no void. Dislocations formed more readily around larger voids.