
Recommended
More Related Content
Similar to Neurodegenrtative disorders.pptx
Similar to Neurodegenrtative disorders.pptx (20)
More from AyeshaSaleem423816
More from AyeshaSaleem423816 (18)
Recently uploaded
Recently uploaded (20)
Neurodegenrtative disorders.pptx
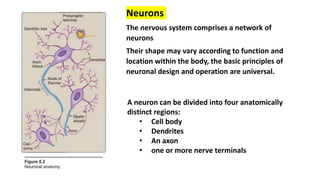
- 1. Neurons The nervous system comprises a network of neurons Their shape may vary according to function and location within the body, the basic principles of neuronal design and operation are universal. A neuron can be divided into four anatomically distinct regions: • Cell body • Dendrites • An axon • one or more nerve terminals
- 2. • Principal functional unit of the central nervous system (CNS) is the neuron. Neurons of different types and in different locations have distinct properties, including diverse functional roles, patterns of synaptic connections, neurotransmitters used, and metabolic requirements, which vary with electrical activity. • Mature neurons are incapable of cell division, so destruction of even a small number of neurons essential for a specific function may produce a neurologic deficit. In addition to neurons, the CNS contains other cells, such as astrocytes and oligodendrocytes, which make up the glia. • Different cellular components of the CNS are affected by distinct unique neurologic disorders and also respond to common insults (e.g., ischemia, infection) in a manner that is distinct from other tissues.
- 3. Neurodegenerative diseases /disorders Neurodegenerative diseases are disorders characterized by the progressive loss of particular groups of neurons, which often have shared functions. Different diseases tend to involve particular neural systems and have relatively stereotypic presenting signs and symptoms. Pathologic process that is common across most of the neurodegenerative diseases is the accumulation of protein aggregates (hence the occasional use of the term “proteinopathy”). Protein aggregates may arise because of mutations that alter the affected protein’s conformation or disrupt the pathways that are involved in the processing or clearance of an otherwise normal protein.
- 4. In other situations, there may be a subtle imbalance between protein synthesis and clearance that allows gradual accumulation of proteins. Protein aggregates typically are resistant to degradation and show aberrant localization within neurons, as more and more protein is shunted into the aggregates, the normal function of the protein may also be lost, and this may also contribute to cell injury. Protein aggregates are capable of behaving like prions, aggregates derived from one cell may be taken up by another and provoke additional protein aggregation. Protein aggregates are recognized histologically as inclusions, which serve as diagnostic hallmarks.
- 5. The basis for aggregation varies from one disease to another. It may be directly related to an intrinsic feature of a mutated protein • Huntington disease [HD]: expanded polyglutamine repeats • Alzheimer disease [AD], in an intrinsic feature of a peptide derived from a larger precursor protein (e.g., Aβ) • Parkinson disease [PD]: Alteration of a normal cellular protein (e.g., α- synuclein Neurodegenerative diseases vary with respect to the anatomic localization of involved areas and their specific cellular abnormalities (e.g., tangles, plaques, Lewy bodies).
- 6. Prion Diseases Prion diseases are rapidly progressive neurodegenerative disorders caused by aggregation and intercellular spread of a misfolded prion protein (PrP); they may be sporadic, familial, or transmitted. Prion diseases include • Creutzfeldt-Jakob disease (CJD), Gerstmann-SträusslerScheinker syndrome, fatal familial insomnia, and kuru in humans • Scrapie in sheep and goats • Mink-transmissible encephalopathy • Chronic wasting disease of deer and elk • Bovine spongiform encephalopathy. All of these diseases are characterized morphologically by “spongiform change” caused by intracellular vacuoles in neurons and glia, and clinically by a rapidly progressive dementia.
- 7. Pathogenesis Prion diseases are caused by “spreading” of misfolded proteins Normal PrP is a 30-kD cytoplasmic protein of unknown function. Disease occurs when PrP undergoes a conformational change from its normal α-helix– containing isoform (PrPsc ) to an abnormal β-pleated sheet isoform, usually termed PrPsc (for scrapie) associated with this conformational change Accumulation of PrPsc in neural tissue seems to be the cause of pathologic changes in these diseases, but how this material induces the development of cytoplasmic vacuoles and eventual neuronal death is still unknown.
- 9. Alzheimer Disease (AD) AD is the most common cause of dementia in older adults, with an increasing incidence as a function of age. The disease usually becomes clinically apparent as insidious impairment of higher cognitive functions. As the disease progresses, deficits in memory, visuospatial orientation, judgment, personality, and language gradually emerge; over a course of 5 to 10 years, the affected individual becomes profoundly disabled, mute, and immobile. Patients rarely become symptomatic before 50 years of age; the incidence of the disease increases with age, and the prevalence roughly doubles every 5 years This progressive increase in incidence with increasing age has given rise to major medical, social, and economic concerns in countries with aging populations. About 5% to 10% of cases are familial https://www.youtube.com/watch?v=v5gdH_Hydes https://www.youtube.com/watch?v=I6K10aif0tE https://www.youtube.com/watch?v=NjgBnx1jVIU
- 10. Pathogenesis Fundamental abnormality in AD is the accumulation of two proteins (Aβ and tau) in specific brain regions, likely as a result of excessive production and defective removal (Fig. 28.35). Two pathologic hallmarks of AD, particularly evident in the end stages of the illness, are amyloid plaques and neurofibrillary tangles. Plaques are deposits of aggregated Aβ peptides in the neuropil, while tangles are aggregates of the microtubule binding protein tau, which develop intracellularly and then persist extracellularly after neuronal death. Both plaques and tangles appear to contribute to the neural dysfunction, and the interplay between the processes that lead to the accumulation of these two types of abnormal protein aggregates is a critically important aspect of AD pathogenesis
- 11. Figure 28.35 Protein aggregation in Alzheimer disease. Amyloid precursor protein cleavage by α-secretase and γ-secretase produces a harmless soluble peptide, whereas amyloid precursor protein cleavage by β-amyloid–converting enzyme and γ- secretase releases Aβ peptides, which form pathogenic aggregates and contribute to the characteristic plaques and tangles of Alzheimer disease. Formation of amyloid plaques found in Alzheimer disease (AD). [Note: Mutations to presenilin, the catalytic subunit of γ- secretase, are the most common cause of familial AD.]
- 12. Role of Aβ. Amyloid precursor protein (APP) is a cell surface protein with a single transmembrane domain The Aβ portion of the protein extends from the extracellular region into the transmembrane domain (Fig. 28.35). Processing of APP begins with cleavage in the extracellular domain, followed by an intramembranous cleavage. If the first cut occurs at the α-secretase site, then Aβ is not generated (the non- amyloidogenic pathway), involved in the shedding of surface proteins. Amyloidogenic pathway: Cleavage by γ-secretase and β-secretase releases Aβ42, which is prone to aggregation and amyloid formation. Once generated, Aβ42 is highly prone to aggregation—first into small oligomers (which appear to be the toxic form responsible for neuronal dysfunction), and eventually into large aggregates and fibrils that can be visualized by microscopy.
- 13. Familial forms of AD support the central role of Aβ generation as a critical step for initiation of AD pathogenesis. Mutations also lead to a gain of function, such that the γ-secretase complex generates increased amounts of Aβ, particularly Aβ42. • Role of tau. Because neurofibrillary tangles contain the tau protein. Tau is a microtubule-associated protein present in axons in association with the microtubular network. With the development of tangles in AD, it shifts to a somatic-dendritic distribution, becomes hyper phosphorylated, and loses the ability to bind to microtubules. The formation of tangles is an important component of AD, and the increased tangle burden in the brain 1. Over aggregates of tau protein elicit a stress response; and 2. Loss of tau protein destabilizes microtubules. https://www.youtube.com/watch?v=vtOKJequi3A
- 14. Parkinson Disease (PD) PD is a neurodegenerative disease marked by a hypokinetic movement disorder that is caused by loss of dopaminergic neurons from the substantia nigra. The clinical syndrome of parkinsonism combines diminished facial expression (often termed masked facies), stooped posture, slowing of voluntary movement, festinating gait (progressively shortened, accelerated steps), rigidity, and a “pill- rolling” tremor. Pathogenesis : PD is associated with protein accumulation and aggregation, mitochondrial abnormalities, and neuronal loss in the substantia nigra and elsewhere in the brain. https://www.youtube.com/watch?v=7upHDhAmkqU
- 15. The first mutations identified as a cause of autosomal dominant PD involved SNCA, a gene that encodes α-synuclein, an abundant lipid-binding protein normally localized to synapses. This protein is a major component of the Lewy body, which is the diagnostic hallmark of PD. The occurrence of disease caused by increases in gene copy number α-synuclein forms aggregates; of these, small oligomers appear to be the most toxic to neurons. There is also evidence that aggregates can be released from one neuron and taken up by another, suggesting a prion-like pattern of spread within the brain
- 16. Huntington Disease (HD) HD is an autosomal dominant disease caused by degeneration of striatal neurons and characterized by a progressive movement disorder and dementia. The disease is relentlessly progressive and uniformly fatal, with an average course of about 15 years. Pathogenesis HD is a prototypic polyglutamine trinucleotide repeat expansion disease. Normal HTT genes contain 6 to 35 copies of the repeat; when the number of repeats is increased beyond this level, it is associated with disease. Repeat expansions occur during spermatogenesis, so that paternal transmission is associated with earlier onset in the next generation, a phenomenon termed anticipation. Expansion of the polyglutamine region bestows a toxic gain-of-function on huntingtin. https://www.youtube.com/watch?v=rcFlo2fDMMs
- 17. Various approaches to silencing expression of the mutant allele are being investigated as potential therapies. Transcriptional dysregulation has been implicated in HD, mutant forms of huntingtin bind various transcriptional regulators. Some of the transcription factors that interact with mutant huntingtin are involved in mitochondrial biogenesis and protection against oxidative injury, and their reduced activity may result in increased susceptibility to oxidative stress. Other alterations that may contribute to the pathogenesis of HD include reduced expression of the growth factor brain-derived neurotrophic factor (BDNF), and disruption of proteasomal and autophagic degradation pathways. Aggregated huntingtin can be transferred between cells, another example of possible prion-like spread of a pathogenic protein
- 18. Neurotrophic factors: Neuropeptides • In the nervous system, neurotrophic factors play a role during development, especially for the differentiation of neuronal and glial cells. They promote cell survival of neurons, axons, and oligodendrocytes, as well as their precursors, in vitro and in lesional paradigms. • The use of neurotrophic factors as therapeutic agents is a novel strategy for restoring and maintaining neuronal function during neurodegenerative disorders Examples of Neurotrphic factors Neurotrophic growth factor (NGF ), Brain-derived neurotrophic factor (BDNF), Neurotrophin-3/4/5 , Ciliary neurotrophic factor, Glial cell line-derived neurotrophic factor
- 19. • Neurotrophic factors (NTFs) can be administered exogenously either as recombinant proteins or via gene delivery. Based on animal models of neurological • NTFs are candidates for preventing neurodegeneration and promoting neuroregeneration. One approach to delivering NTFs to diseased neurons is through viral vector-mediated gene delivery. Viral vectors are now routinely used as tools for studying gene function as well as developing gene-based therapies for a variety of diseases. Transplantation of neural stem/precursor cells (NPCs) has been proposed as a promising therapeutic strategy in almost all neurological disorders characterized by the failure of central nervous system (CNS) endogenous repair mechanisms in restoring the tissue damage and rescuing the lost function.
- 20. Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that typically results in death within 3–5 years after diagnosis. To date, there is no curative treatment and therefore an urgent need of neuroprotective and/or neurorestorative treatments. Due to their spectrum of capacities in the central nervous system—e.g., development, plasticity, maintenance, neurogenesis—neurotrophic growth factors (NTF) have been exploited for therapeutic strategies in ALS Gene therapy has the potential to provide therapeutic benefit to millions of people with neurodegenerative diseases through several means, including Direct correction of pathogenic mechanisms Neuroprotection Neurorestoration Symptom control.
- 22. The chronic nature of many diseases affecting the central nervous system makes gene therapy an attractive alternative treatment option. Delivery of therapeutic molecules to the CNS has always been a significant challenge due to the physical barriers imposed by both the skull and the blood– brain barrier, which prevents the passage of large molecules from the bloodstream into brain tissue. The brain is composed of both neurons and non-neuronal cell types. Cell-to-cell communication occurs locally but also via interconnecting circuits throughout the brain. Moreover, there is global involvement of the brain in certain diseases (e.g. AD) but the involvement of very specific regions in other diseases (e.g. PD).
- 23. There are still several hurdles to overcome such as the refinement and incorporation of regulatory systems into gene cassettes for safety purposes and development of vector systems capable of widespread global transduction of large brain regions for certain neurological diseases. This field is still in its early days, but data from early clinical trials have shown promising outlooks for this technology, paving the way for more widespread evaluation of the technology in humans Neural transplantation and stem cells of neural stem/precursor cells (NPCs) has been proposed as a promising therapeutic strategy in almost all neurological disorders characterized by the failure of central nervous system (CNS) endogenous repair mechanisms in restoring the tissue damage and rescuing the lost function.
- 24. Spinal cord injury (SCI) • Traumatic spinal cord injury (SCI) is a major cause of disability. The financial burden is significant in terms of direct health care costs as well as loss of economic productivity. Even small improvements in mobility and/or manual dexterity may substantially reduce these costs and improve quality of life . • Current therapeutic options remain limited, and there is a need for more effective treatments to restore function in SCI patients. • Cellular therapy with intravenous infusion of mesenchymal stem cells (MSCs) derived from bone marrow improves functional outcome in experimental models of SCI. • While the mechanisms underlying these beneficial effects have not been fully elucidated
- 25. Potential mechanisms include • Neuroprotection and immunomodulation • Induction of axonal sprouting • Remyelination • Restoration of blood-brain/spinal cord barrier • Enhancement of remote gene expression responses in brain. Several clinical trials for SCI using autologous cultured MSCs derived from bone marrow have been reported. MSCs in these studies are delivered via • Intramedullary injection • Intrathecally • Intravenously
- 26. Honmou et al., 2021 used Intravenous injection of bone marrow derived stem cells (MSCs) in patients with spinal cord injuries and it led to significant improvement in motor functions. Their observations support safety, feasibility and provide initial data that suggests rapid functional improvements following MSC infusion. For more than half of the patients, substantial improvements in key functions — such as ability to walk, or to use their hands — were observed within weeks of stem cell injection. No substantial side effects were reported. Osamu Honmou , Toshihiko Yamashita , Tomonori Morita , Tsutomu Oshigiri , Ryosuke Hirota , Satoshi Iyama , Junji Kato , Yuichi Sasaki , Sumio Ishiai , Yoichi M. Ito, Ai Namioka , Takahiro Namioka , Masahito Nakazaki , Yuko Kataoka-Sasaki , Rie Onodera, Shinichi Oka, Masanori Sasaki, Stephen G. Waxman, Jeffery D. Kocsis. 2021. Intravenous infusion of auto serum-expanded autologous mesenchymal stem cells in spinal cord injury patients: 13 case series. Clinical Neurology and Neurosurgery. 203 (2021) 106565.
- 27. Stem cells can potentially provide trophic support to the injured spinal cord microenvironment by modulating the inflammatory response, increasing vascularization and suppressing cystic change. Bone marrow-derived mesenchymal stem cells. BMSC is the most popular source of MSCs for the treatment of SCI with more than 20 clinical trials registered on the Clinical Trials The patients had sustained non-penetrating spinal cord injuries, in many cases from falls or minor trauma, several weeks prior to implantation of the stem cells. Their symptoms involved loss of motor function and coordination, sensory loss, as well as bowel and bladder dysfunction. The stem cells were prepared from the patients’ own bone marrow, via a culture protocol that took a few weeks in a specialized cell processing center. The cells were injected intravenously in this series, with each patient serving as their own control.
- 28. Injury to the adult mammalian spinal cord results in extensive axonal degeneration, variable amounts of neuronal loss, and often severe functional deficits. Restoration of controlled function depends on regeneration of these axons through an injury site and the formation of functional synaptic connections. One strategy that has emerged for promoting axonal regeneration after spinal cord injury is the implantation of autologous Schwann cells into sites of spinal cord injury to support and guide axonal growth. Further, more recent experiments have shown that neurotrophic factors can also promote axonal growth, and, when combined with Schwann cell grafts, can further amplify axonal extension after injury. Continued preclinical development of these approaches to neural repair may ultimately generate strategies that could be tested in human injury. Leonard L Jones,Martin Oudega, Mary Bartlett Bunge, and Mark H Tuszynski. 2001. Neurotrophic factors, cellular bridges and gene therapy for spinal cord injury. J Physiol. 2001 May 15; 533(Pt 1): 83–89. doi: 10.1111/j.1469-7793.2001.0083b.x
Editor's Notes
- Figure 28.33 Pathogenesis of prion disease. α-helical PrPc may spontaneously shift to the β-sheet PrPsc conformation, an event that occurs at a much higher rate in familial diseases associated with germline PrP mutations. PrPsc may also be acquired from exogenous sources, such as contaminated food, medical instrumentation, or medicines. Once present, PrPsc converts additional molecules of PrPc into PrPsc through physical interaction, eventually leading to the formation of pathogenic PrPsc aggregates.
- Flore Gouel, Anne-Sophie Rolland, Jean-Christophe Devedjian, Thierry Burnouf and David Devos Past and Future of Neurotrophic Growth Factors Therapies in ALS: From Single Neurotrophic Growth Factor to Stem Cells and Human Platelet Lysates. MINI REVIEW article . Front. Neurol., 02 August 2019 | https://doi.org/10.3389/fneur.2019.00835