"O you whohave believed, seek help through patient
& prayer . Indeed, ALLAH is with the patient,"
• Muhammad Naeem Ahmad
• Roll No SSPR139F20
• Session 2020-2022
• Supervisor Professor Dr.Shahzad Naseem
3.
Calculation of Opticaland electronic properties
of ZnCdS thin film using DFT.
•MASTER OF PHYLOSOPHY IN SOLID STATE
PHYSICS
•CENTER OF EXCELLENCE IN SOLID STATE
PHYSICS
•UNIVERSITY OF THE PUNJAB , QUAID-E-AZAM
•CAMPUS LAHORE , PAKISTAN
4.
Introduction
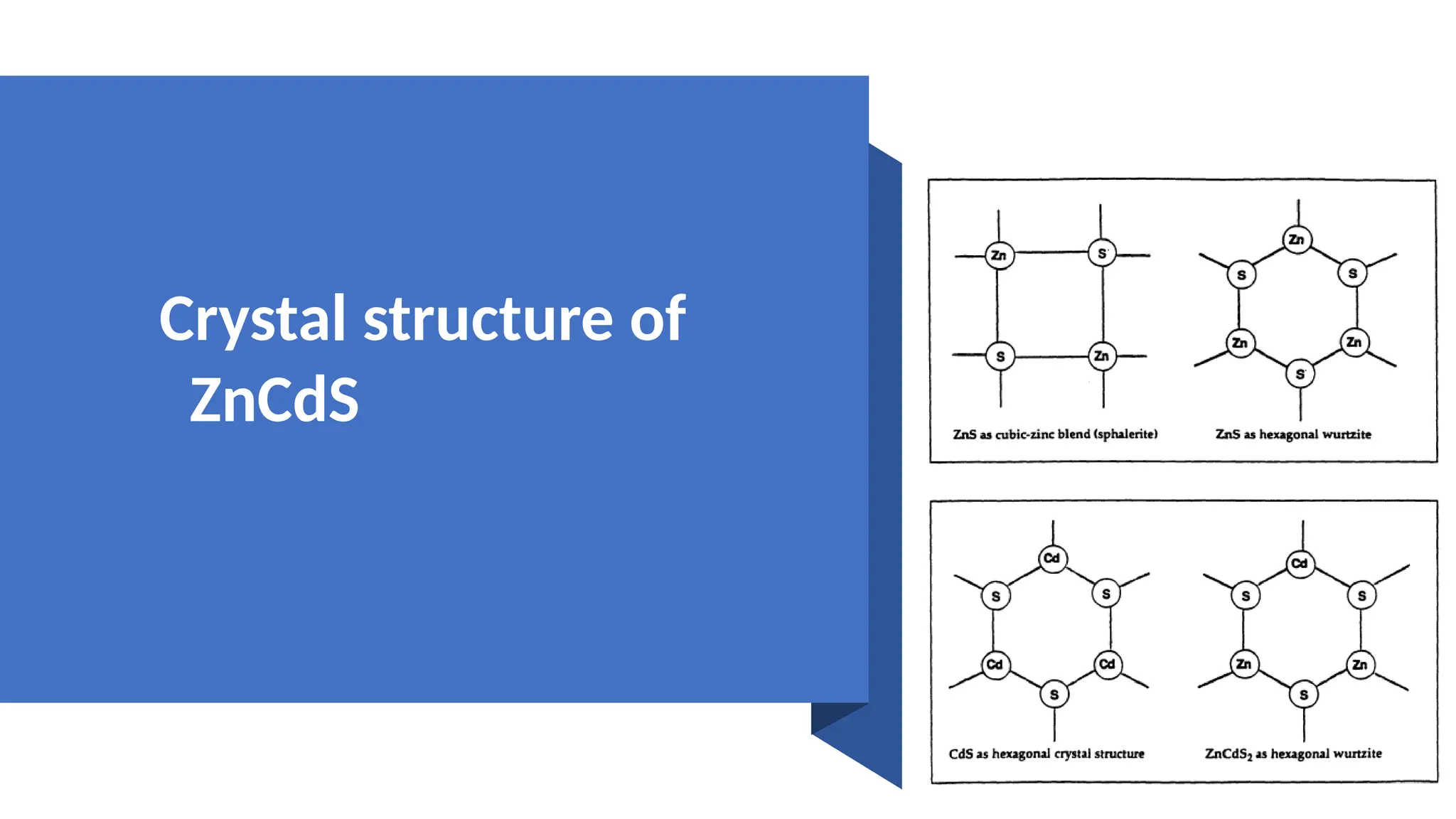
Crystal structure ofZnCdS
rh
Applications of ZnCdS
Literature Review
Experimental and Theoretical Division
Theoretical work (DFT)
Presentation
Outlines
LDA & GGA Results
LDA & GGA Results
5.
Objective • Todevelop highly efficient and
cost effective ZnCdS thin film for
solar cell applications.
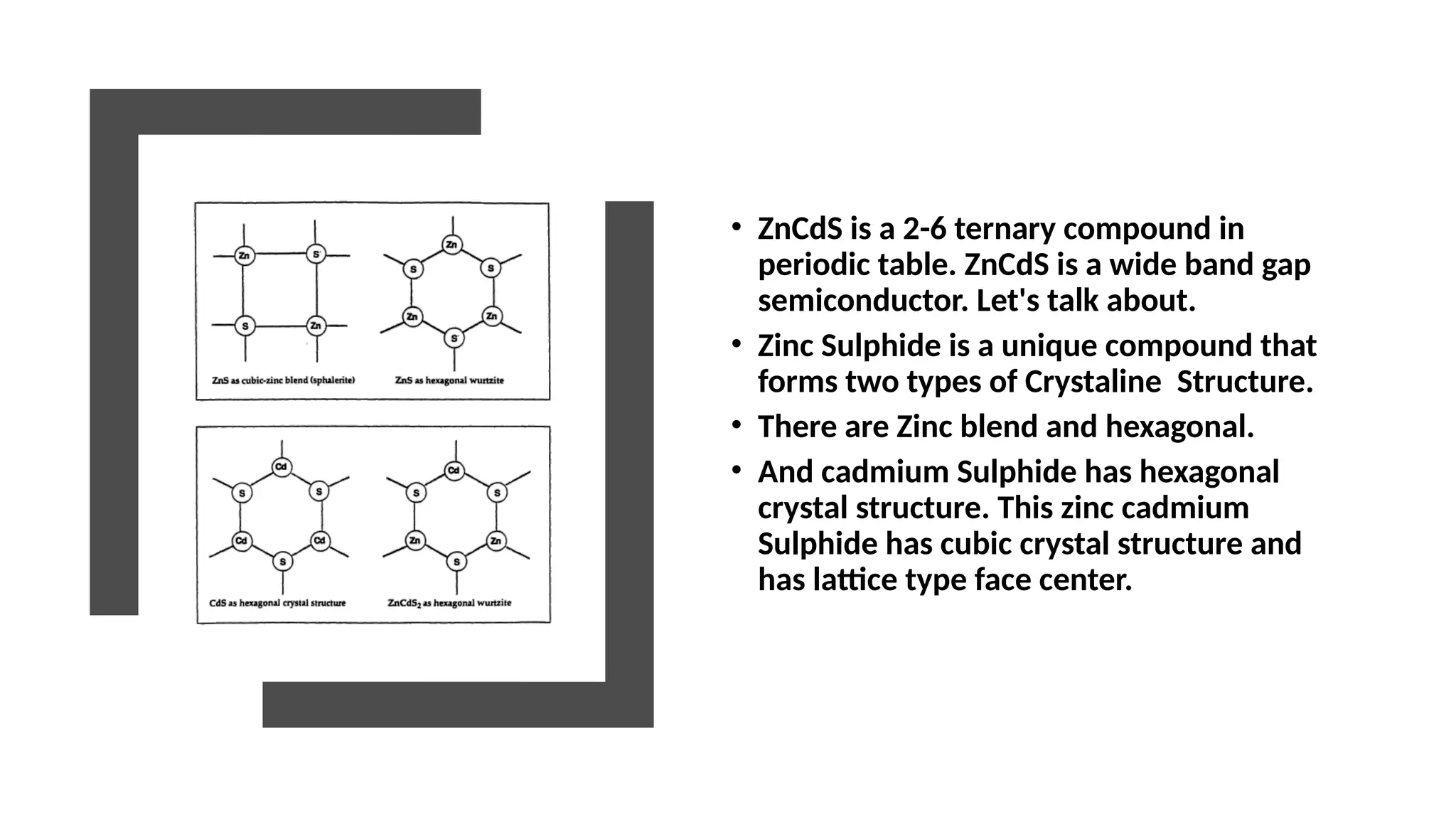
• ZnCdS isa 2-6 ternary compound in
periodic table. ZnCdS is a wide band gap
semiconductor. Let's talk about.
• Zinc Sulphide is a unique compound that
forms two types of Crystaline Structure.
• There are Zinc blend and hexagonal.
• And cadmium Sulphide has hexagonal
crystal structure. This zinc cadmium
Sulphide has cubic crystal structure and
has lattice type face center.
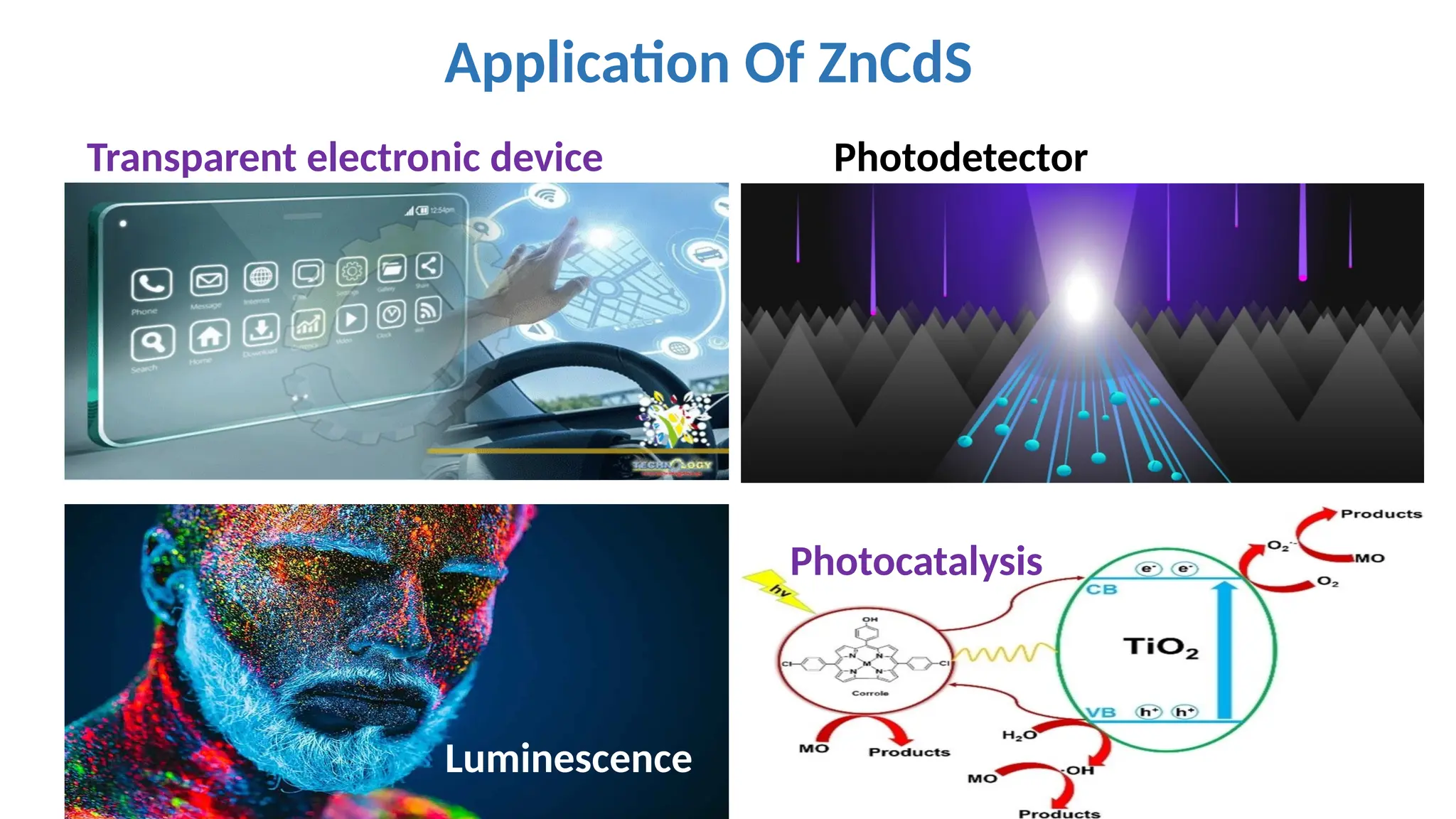
•In general ZnCdSapplication
are in transparent electronic
devices use as photo detector,
for medical diagnosis ,
Car( night vision) in
luminescence and
photocatalytic and mainly in
Soler cell application.
10.
Literature Review
Noor, Naveed
Ahmad
•Electronic structure, density of state and
energy band gap for ZnxCd1-xS is estimated
in the range 0<X<1 using both the standard
local density approximation (LDA) as well as
Generalized gradient approximation (GGA).
• It is observed that the direct band gap Eg of
CdZnS is decreases nonlinearly with the
compositional parameters X, as observed
experimentally it is also found that Cds and
d, S p and Zn d state play a major role in
determining the electronic properties of this
alloy system.
Researcher year
2015
Finding/ conclusion
11.
Theoretical Investigation
• DensityFunction theory (DFT)
• Local density Approximation (LDA)
• Generalized Gradient Approximation (GGA)
• Results
• Conclusions
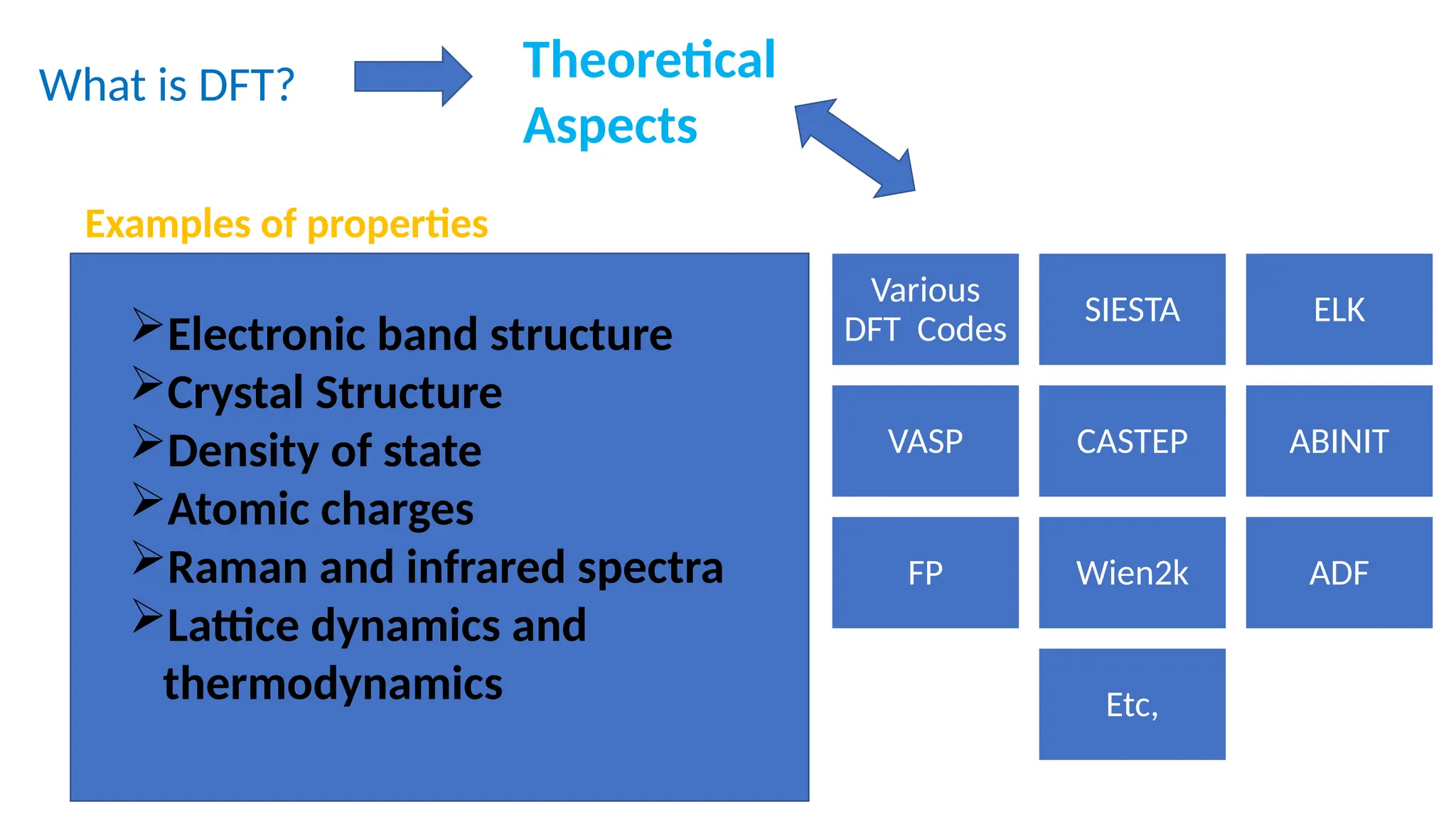
What is DFT?Theoretical
Aspects
Examples of properties
Electronic band structure
Crystal Structure
Density of state
Atomic charges
Raman and infrared spectra
Lattice dynamics and
thermodynamics
Various
DFT Codes
SIESTA ELK
VASP CASTEP ABINIT
FP Wien2k ADF
Etc,
14.
Why Density FunctionTheory
• The calculation of physical and chemical properties of multi- particle systems (atoms,
molecules or solids) require the exact determination of electronic structure and total
energy of these systems
• Schrodinger equation successfully explains the electronic structure of simple systems
and numerically exact solutions are found for small no. of atoms and molecules
• This n-electron problems was solved when Kohn and Sham in 1965 formulated a theory
concerning 3-dimensional electron density and energy functionals.
• Electron density n(r) plays central role instead of wave function. The problem of many
interacting particles system in static potential is reduced to non-interacting single
particle system in an effective potential.
15.
Many body problem:
For large interacting system , we first need to consider a many particle wave function.
Many body Hamiltonian for electron and nucleus is of the form given below
16.
Approximation for solvingmany body problem
The Bom-Oppenheimer approximation
Hartree approximation
Hartree-Fock method
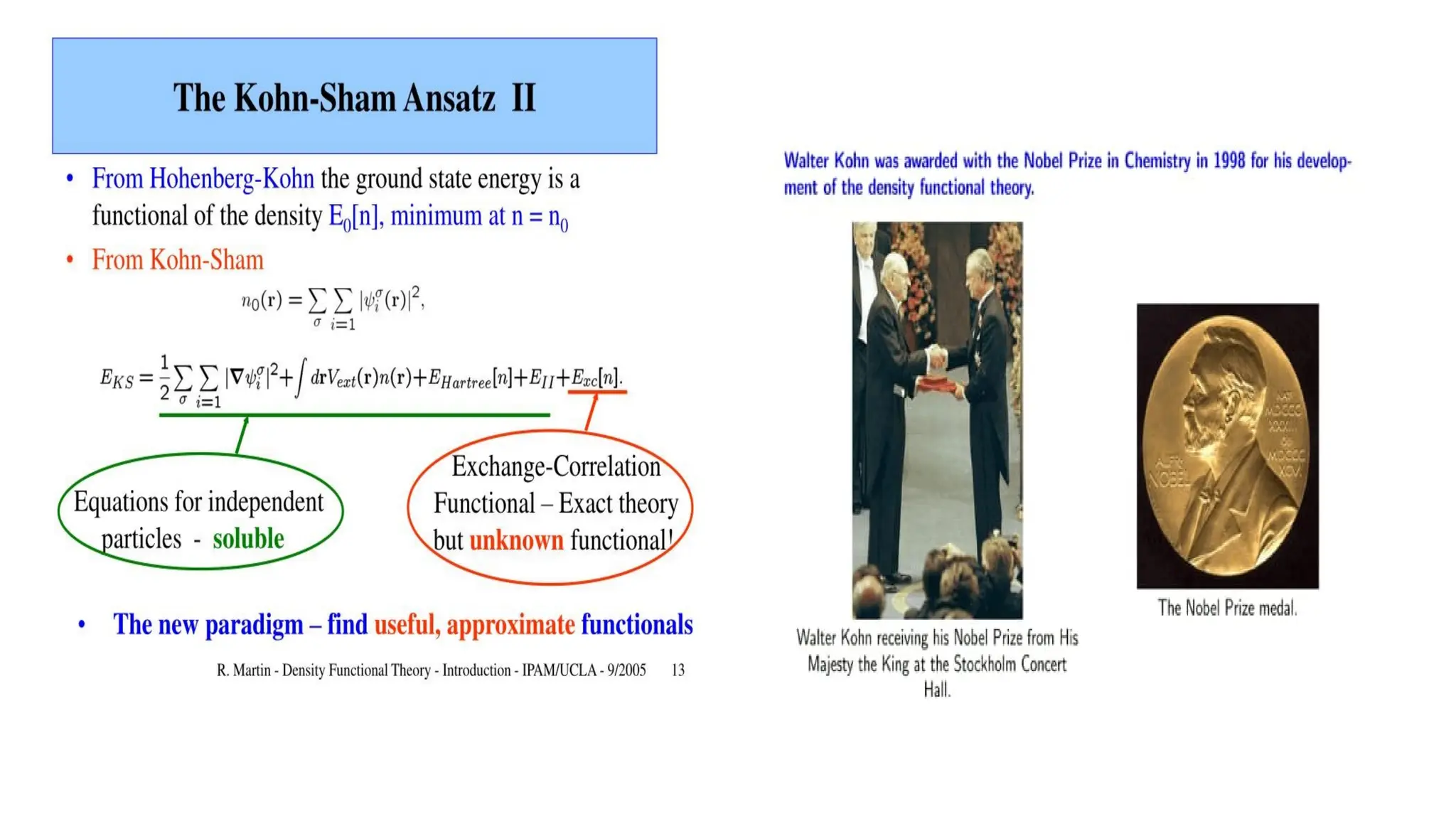
Hohenberge-Kohen
Kohn-Sham approximation ( walter Kohn
and Lu J Sham )
The Nuclei are much heavier than electron.
They move much more slowly and
hence neglect the nuclear kinetic energy.
The wave function separated into electronic and
nuclear part and determine motion of electron with
nuclei held fixed.
1. Borm-Oppenheimer approximation
1927
Max Born and Robert Oppenheimer
17.
Reduce thecomplexity of electron-electron interaction.
Electron are independent and interacts with other in an average way.
For an n-electron system , each electron does not recognize other as
single entities but as a mean field.
Hence, n-electron system becomes a set of interacting one-electron
system where each electron moves in the average density of rest
electrons.
Hartree approximation one electron model
R=Nuclear
r =electron
Vext = electron and
nuclei interaction
potential
VH = Hartree potential
( e-e interaction )
Hartree
1928
18.
Hartree Fock method
Basedon the one-electron and mean field approach by Hartree, V.A. Fock enhanced the method to higher
Perfection . Fock and J.C Slater in 1930 generalized the Hartree's theory to take into account the antisymetric
requirement.
In HF method , the n-electron wave function approximation as a linear combination of non-interacting one-
electron wave function in the form of slater deteminet.
Fock
1930
Difficulties with Hartree-Fock Theory:
Correlation energy
Problem of dealing 3N dimensional.
19.
A new approachhas been develop known as
Density Functional Theory ( DFT )
In 1964 Hohenberg and Kohn showed that Schrodinger equation (3N dimensional e.g
10 electron require 30 dimensions could be reformulated in term of electron density
n(r) with non-interacting n separate 3-dimentional ones.
The main objective of DFT is to replace the many-particle electronic
wavefunction with the electron density as the basic quantity.
The electron density n(r), the central player in DFT decides everything in an N-
electron quantum state where there is no individual electron density but a
3-dimensional of electron.
The addition of all the electron densities over the whole space naturally return to the
total number of electron in the system.
The knowledge of overlapping of atomic electron density , roughly generate
the electron density of the solids.
This theory gives approximate solution to both Exchange and Correlation energies.
20.
The Fundamental Pillarsof DFT
First Hohenberg Kohn ( HK) theorem: The ground-state energy
Is a unique functional of the electron density n(r).
This theorem provide one to one mapping between ground state wave
function and ground state charge density.
The ground state charge density can uniquely describe all the ground
state properties of system.
The fundamental concept behind density functional theory is
that charge density (3-Dimensional) can correctly describe the
ground state of N-particle instead of using a wave function (3N-
Dimensional).
Second Hohenberg Kohn (HK) theorem : The electron density that minimize
the energy of the overall functional is the true electron density:
If the true functional form of energy in term of density gets known, then
one could vary the electron density until the energy from the functional
is minimized , giving us required ground state density.
This is essentially a variational principle and is used in practice with
approximation forms of the functional.
The simplest possible choice of a functional can
be constant electron density all over the space.
Two Hohenberg and Kohn
theorems
The existence of a unique
functional.
The variational principle.
22.
Exchange-correlation application
1. Localdensity approximation ( LDA)
Approximation used to fixed out exchange-
correlation function
Exchange-Correlation energy functional is
purely local.
Ignores corrections to the exchange-
correlation energy at a point r
due to nearly inhomogeneities in the
electron density.
2. Generalized Gradient Approximation (GGA)
Depends on local density and its gradient.
GGA uses information about the local electron density and
also the local gradient in the electron density.
Though GGA includes more physical information than LDA it
is not necessary that it must be more accurate
There are large number of GGA functional depending on
the ways in which information from the gradient of
the electron density can be included in a GGA functional.
DFT in Practice: The exchange-correlation
Functional
The major problem with DFT is that the exact
functionals for exchange and correlation are not
known, except for the free electron gas.
However , approximation exist which permit
the calculation of certain physical quantities
quite accurately. One of them is LDA.
The local spin-density approximation (LSDA) is a
straightforward generalized of the LDA to
Include electron spin
After LDA underestimate of exchange energy and
overestimate of correlation energy. GGA solve this
problem by adding gradient function
Amsterdam Density Function(ADF)
ADF excels in spectroscopy , transition metals, and heavy element problems.
A periodic structure counterpart of ADF named BAND is available
to study bulk crystals, polymers and surfaces.
ADF offers unique capabilities to predict molecular properties of nanoparticles and
organic electronics materials.
ADF is particularly strong in understanding structure reactivity and spectra of molecules.
DFT calculation are easily prepared and analysed with integrated graphical user interface.
34.
Computational Details
Structure isbuilt for cubic group.
Space Group is Fm3m
Lattice parameter a = 5.41A0
Calculations are carried out using Local Density Approximation (LDA)
We have also used Generalized Gradient Approximation (GGA) for calculations.
35.
Comparison and conclusion
ØZnCdS have been successfully synthesized via thermal evaporation deposition technique and using
Density functional theory via ADF code
Ø To analyse the optical properties of the thin film we use UV-Visible spectrophotometer.
Ø Transmission spectra shows almost 90% transmission.
Ø ZnCdS films show good transmission in the visible region so finds the best use as Buffer/window layer.
Ø Optical direct band gap is evaluated to be 4.08eV and theoretically obtained band gap is 3.640eV.
Ø The theoretical and experimental value of band gap are about in agreement.
Ø For both approximation ( LDA and GGA), the density of state is discontinus for the width from the top of the
valence band to the bottom of the conduction band which is normally the bandgap of the system.
Ø There was no effect of functional change we got same values.
#22 The LDA assumes that the density is the same everywhere. Because of this, the LDA has a tendency to underestimate the exchange energy and over-estimate the correlation energy.[24] The errors due to the exchange and correlation parts tend to compensate each other to a certain degree. To correct for this tendency, it is common to expand in terms of the gradient of the density in order to account for the non-homogeneity of the true electron density. This allows corrections based on the changes in density away from the coordinate. These expansions are referred to as generalized gradient approximations (GGA)[25][26][27] and have the following form. Last equation this page
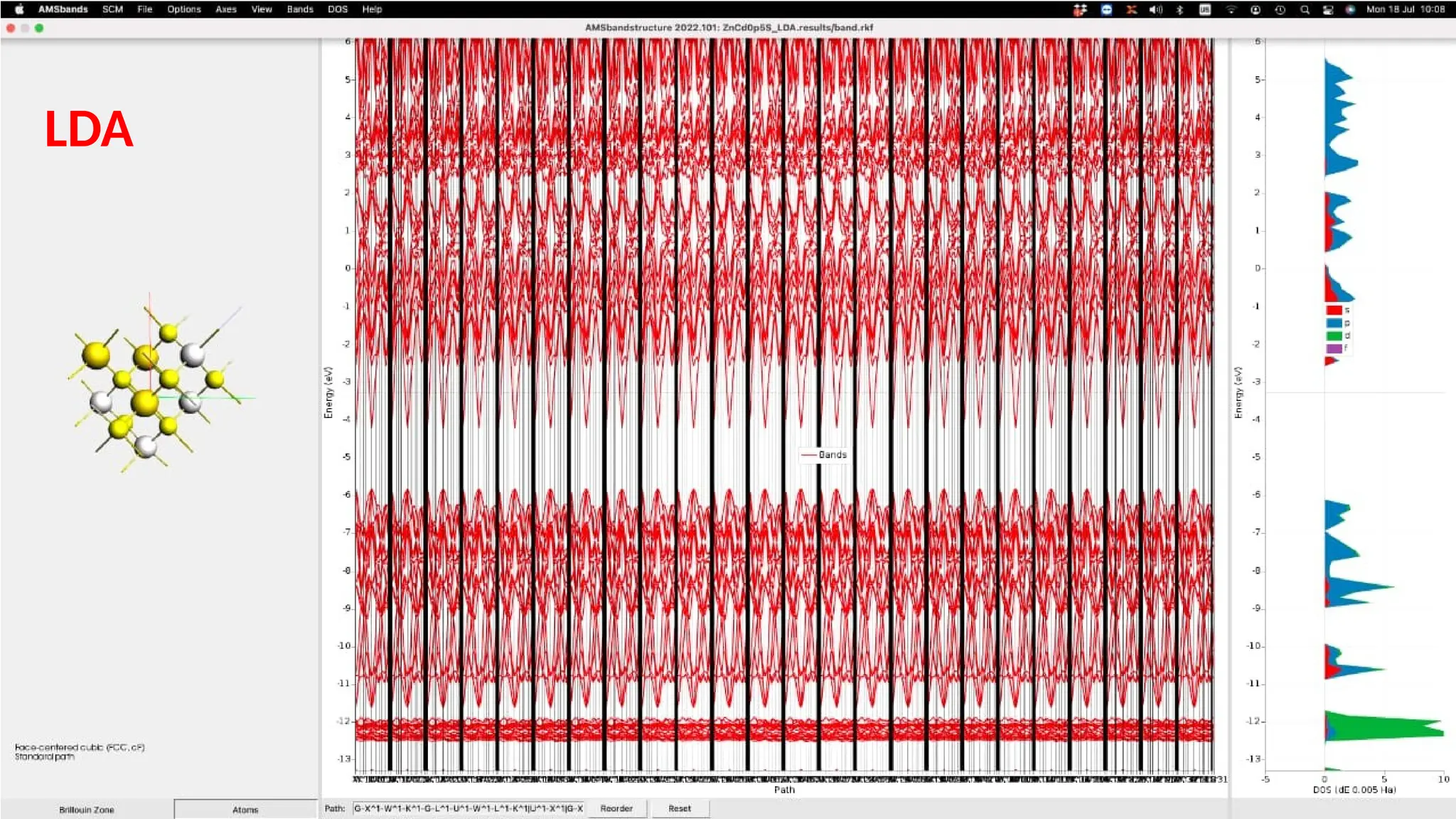
#23 This is the band structure of ZnCdS small structure using 16 atom in the unit cell. The unit cell volume came out to be 316.68084 Angstrom cube. As we can see that the fermi level is at around –3.3eV and there is a huge energy gap b/w the valence and conduction bands. However , there are some bands crossing the Fermi level into the energy gap region.The band gap energy calculated using this band structure is 1.6eV.
The energy band gap calculated with LDA approximation through the density of state plot came out to be 3.64eV as seen on the right side where we can see the density of states plote for s,p,d and f orbitals. It is obvious that there is band in the DOS plot too. The fermi level is seen to be closed.
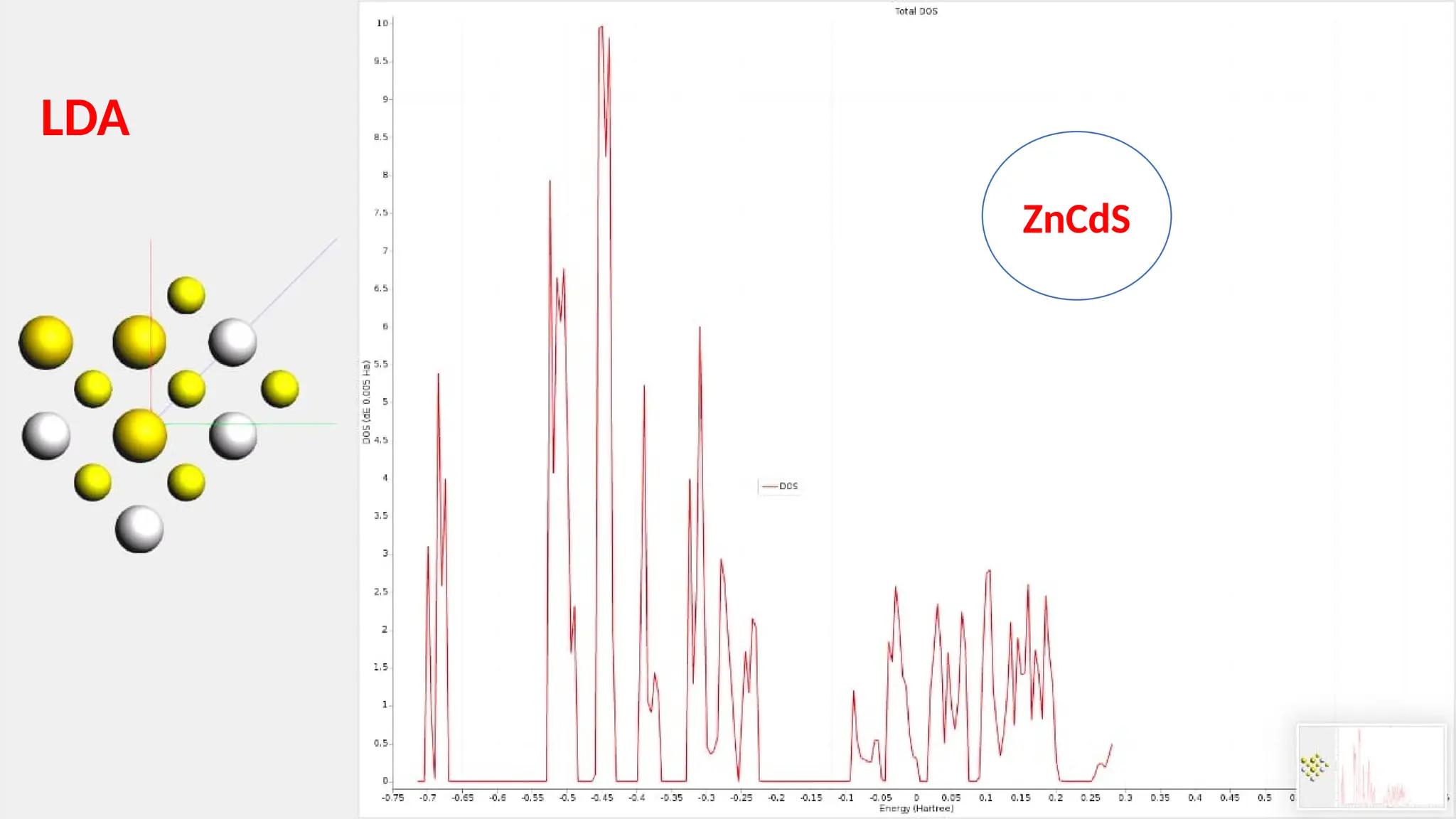
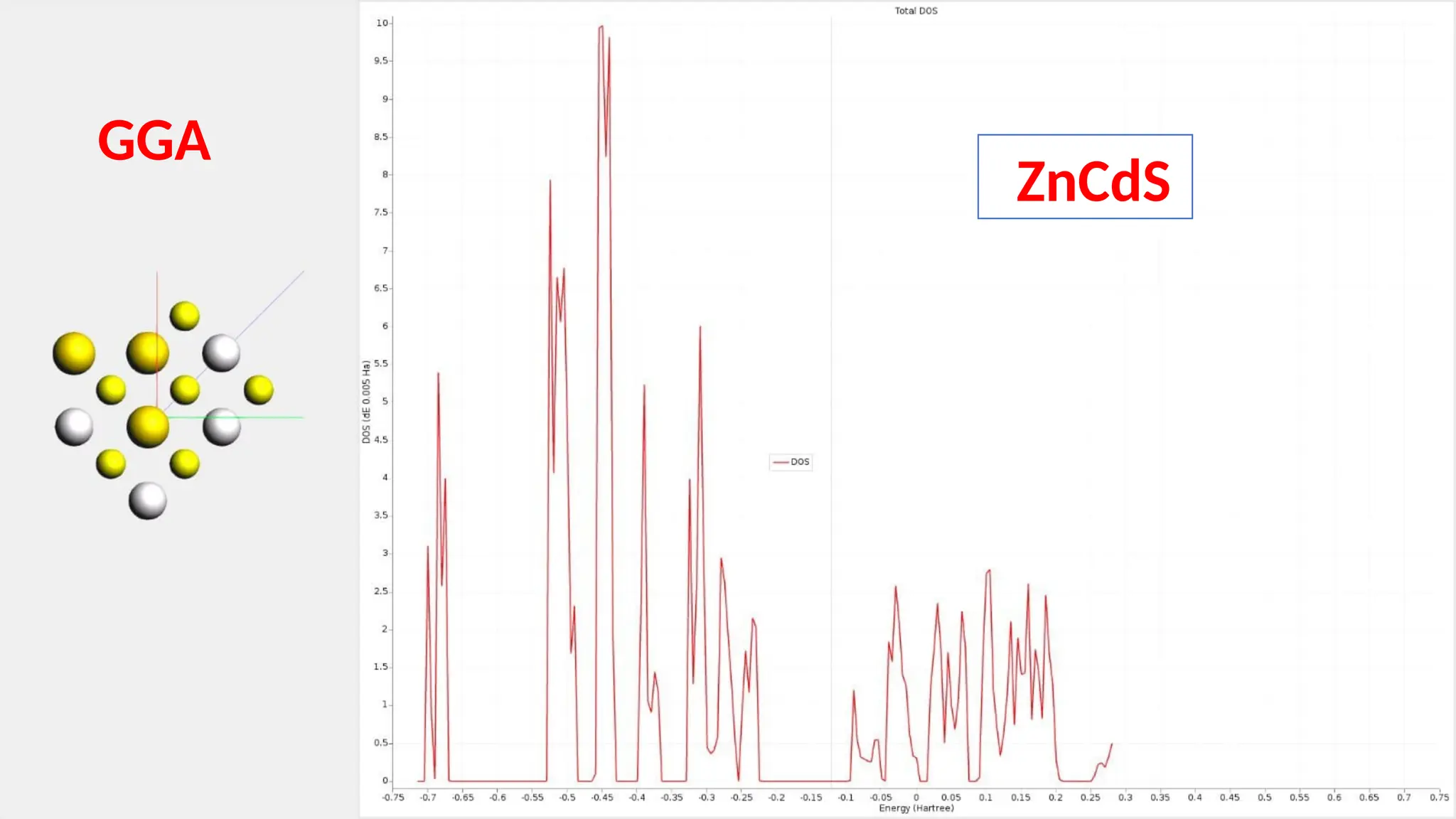
#24 The total density of state plot is shown here in detail. This one again shows an n-type semiconductor with a large band gap value of 3.64eV.
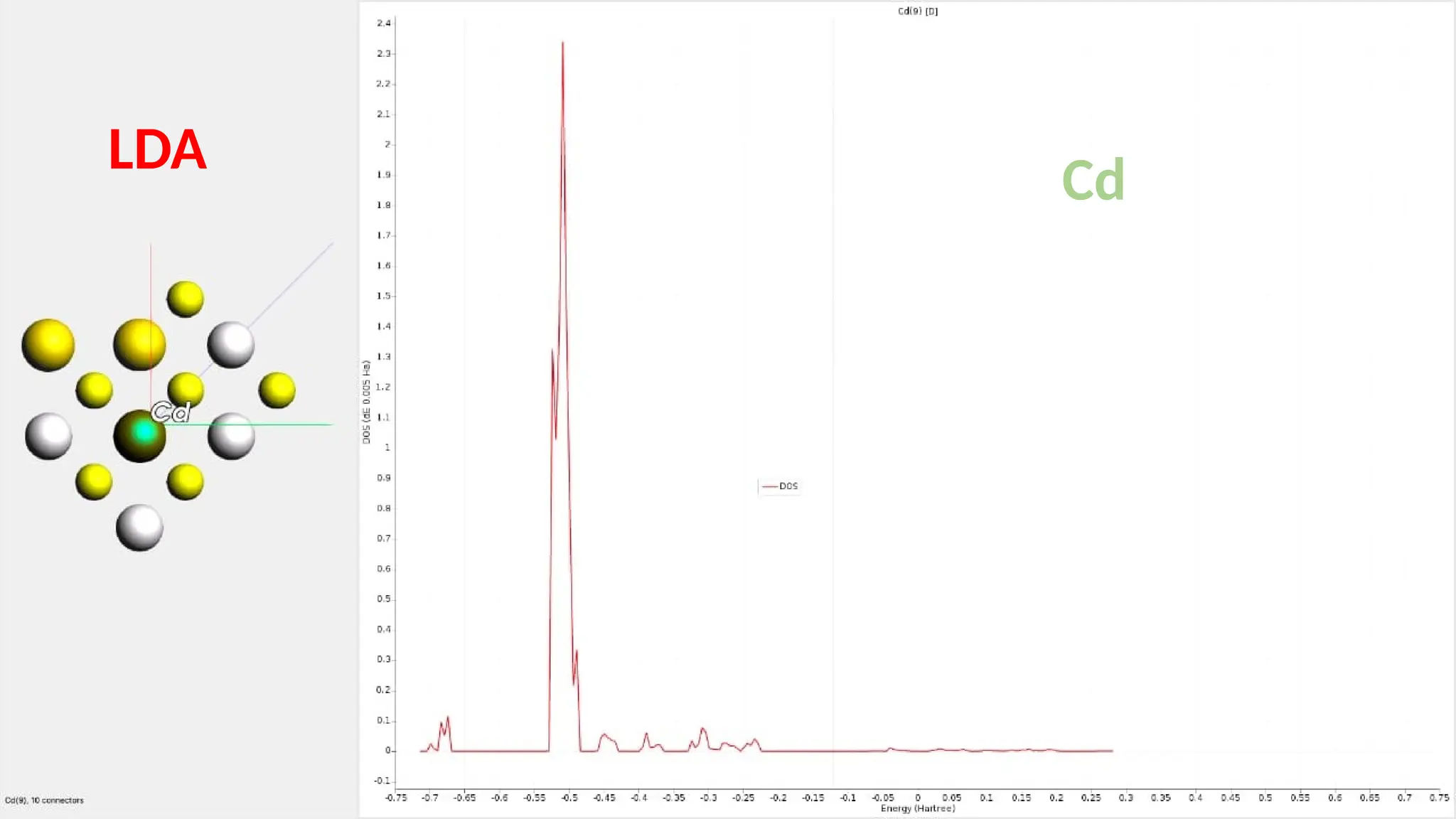
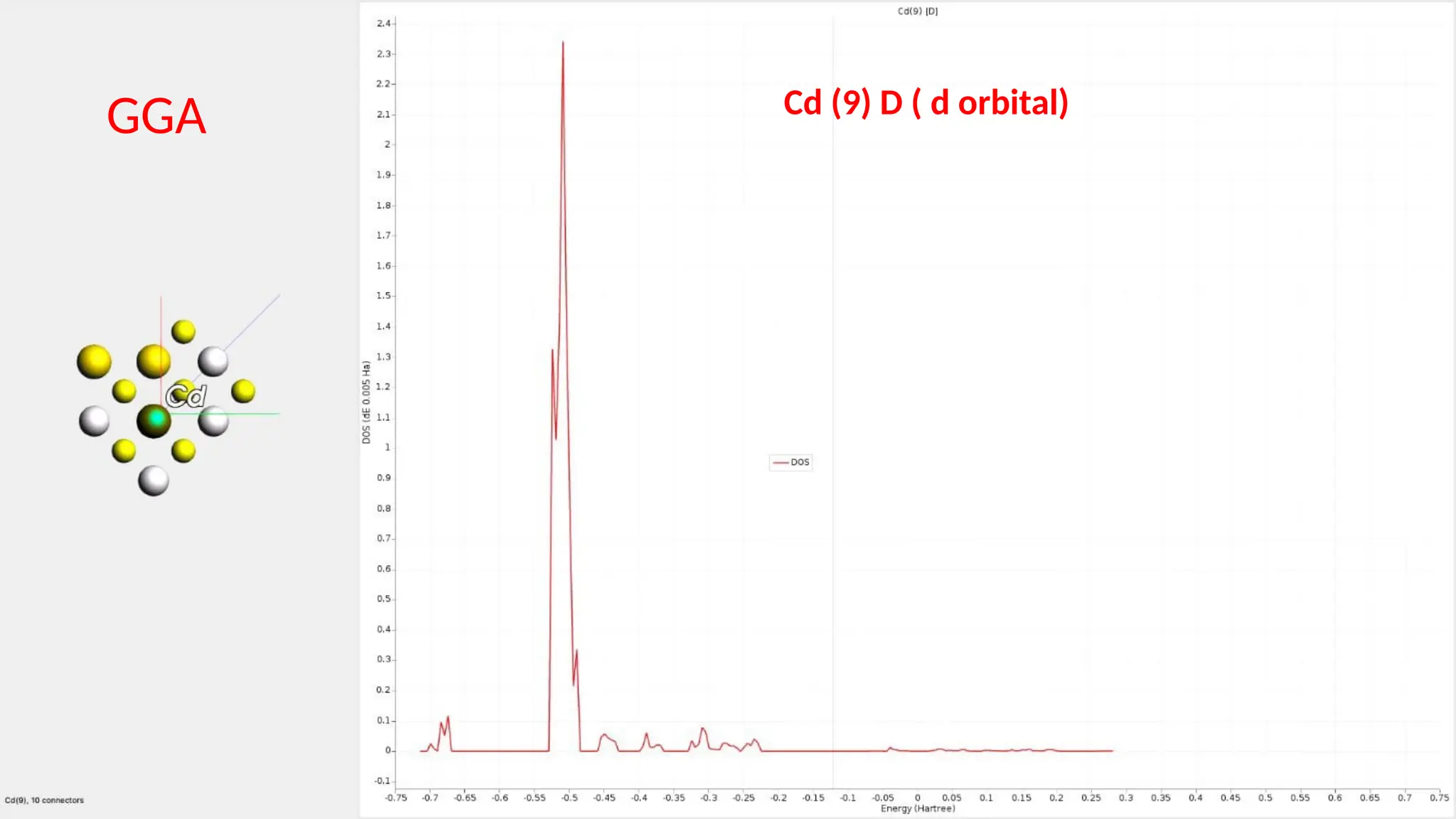
#25 Here are partial density of state for Cd s ,p and d orbitals and it can be seen that the major contribution is of Cd d orbitals.
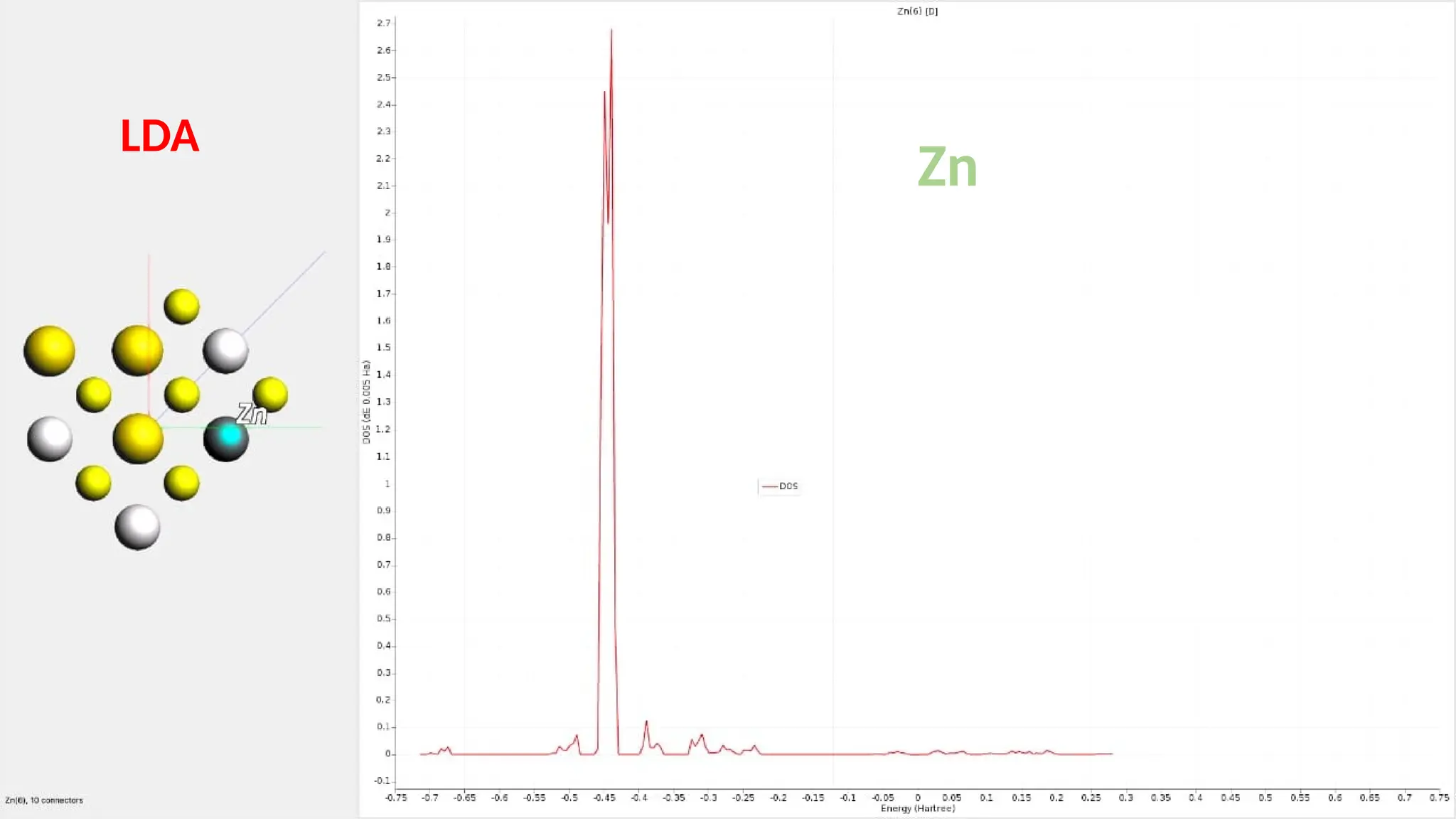
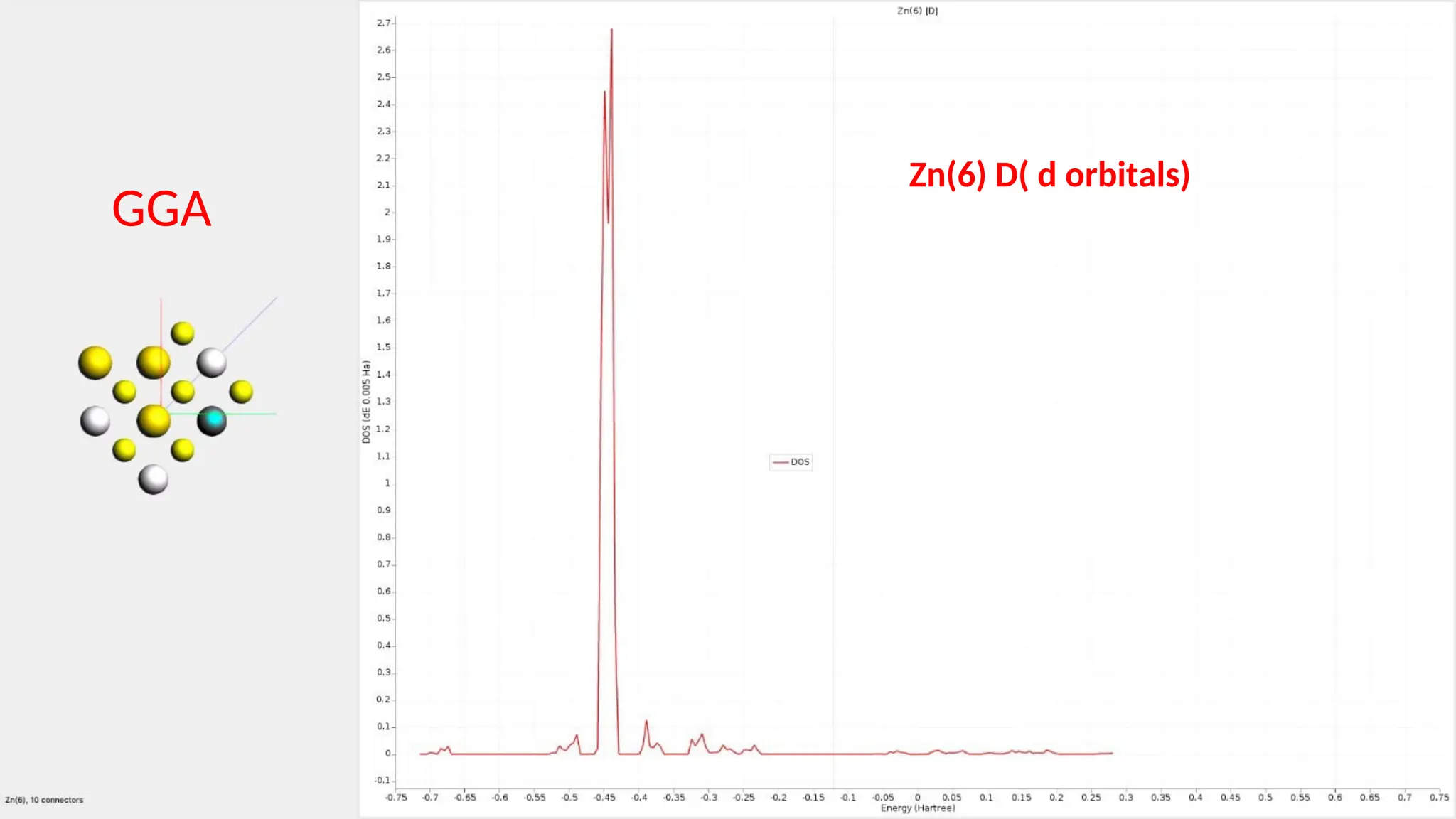
#26 Here are partial density of state for Zn s ,p and d orbitals and it can be seen that the major contribution is of Zn d orbitals.it has 10 connection.
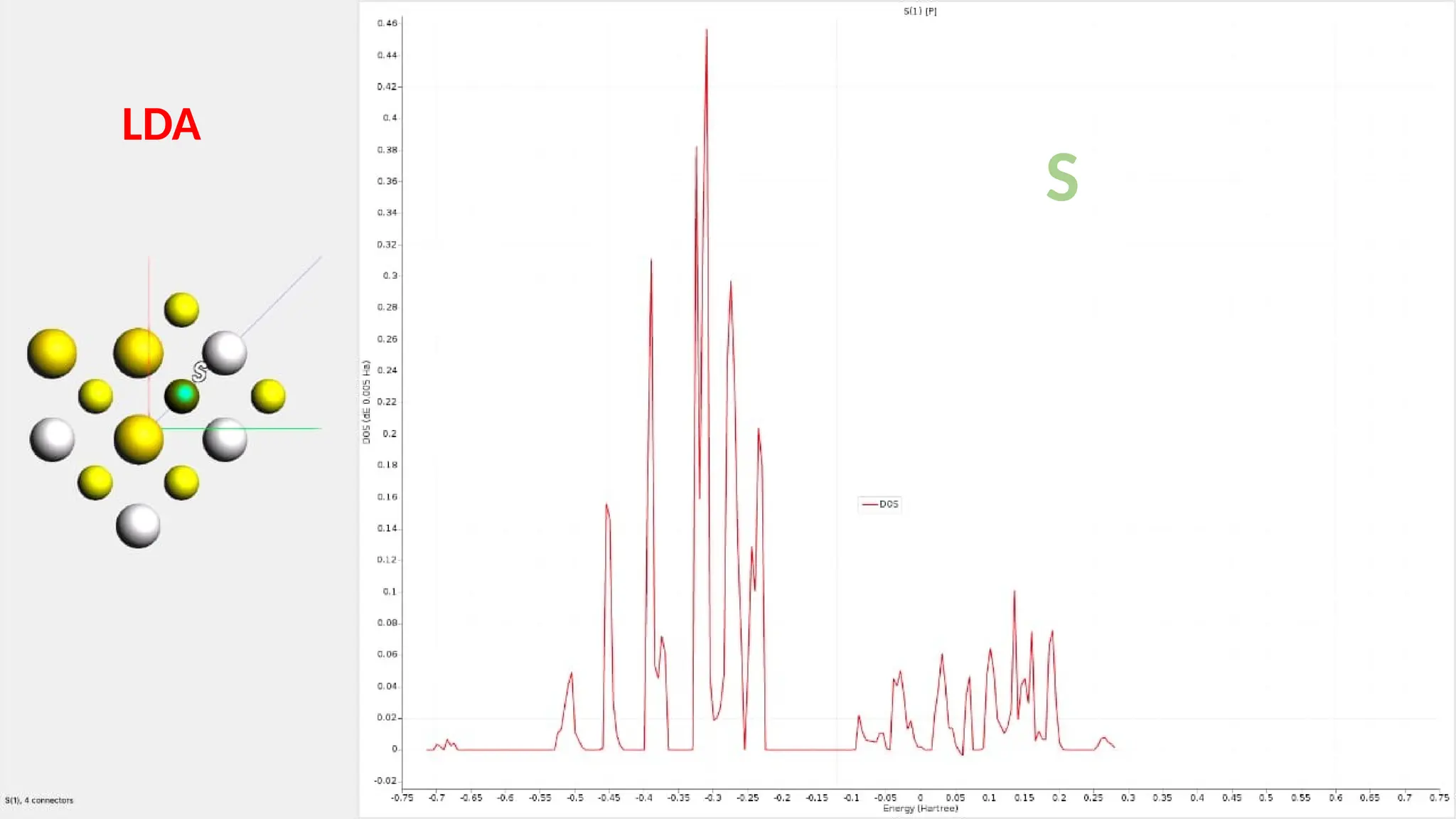
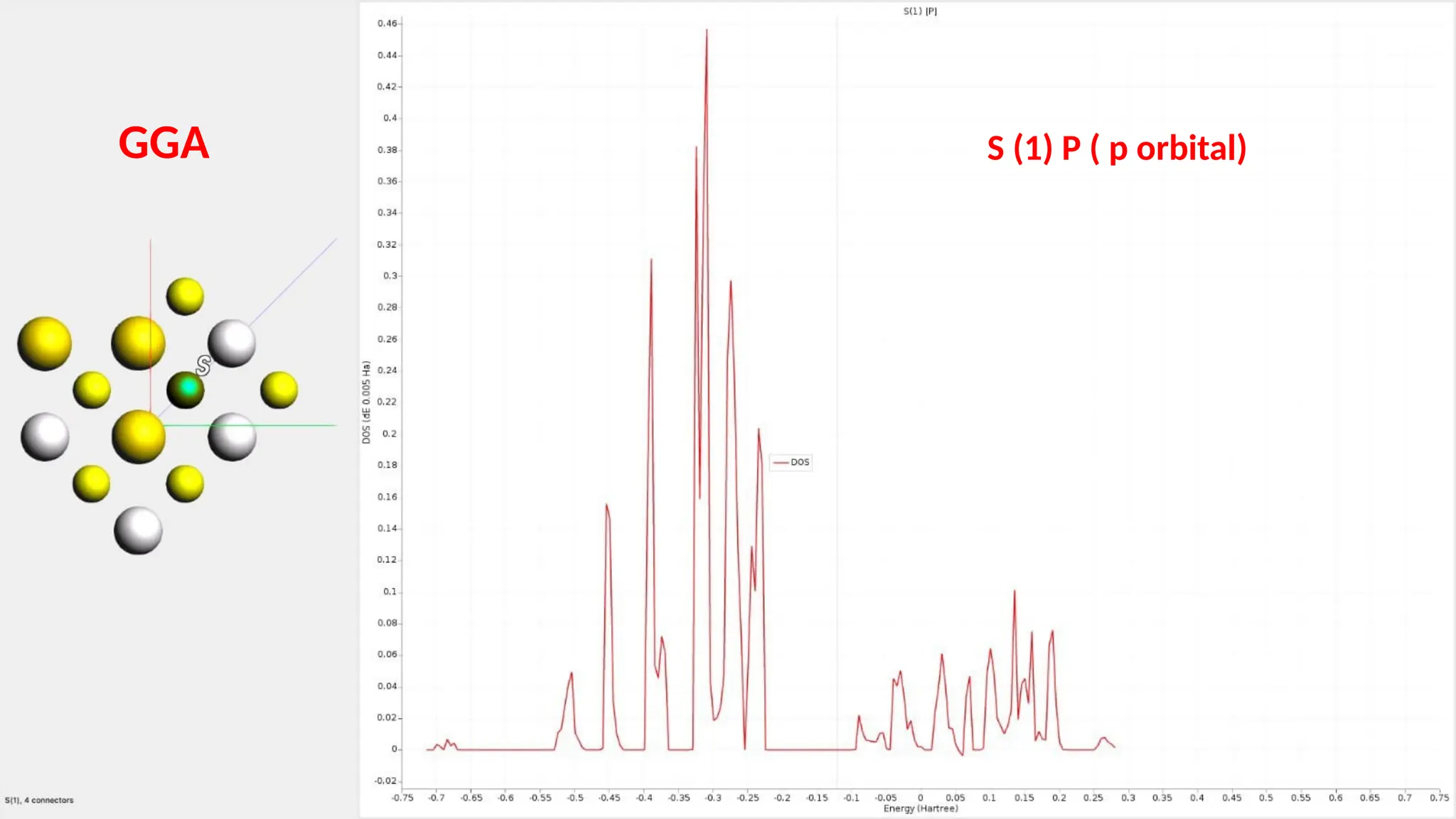
#27 Here are partial density of state for S s ,p and d orbitals and it can be seen that the major contribution is of S p orbitals.it has 4 connection.
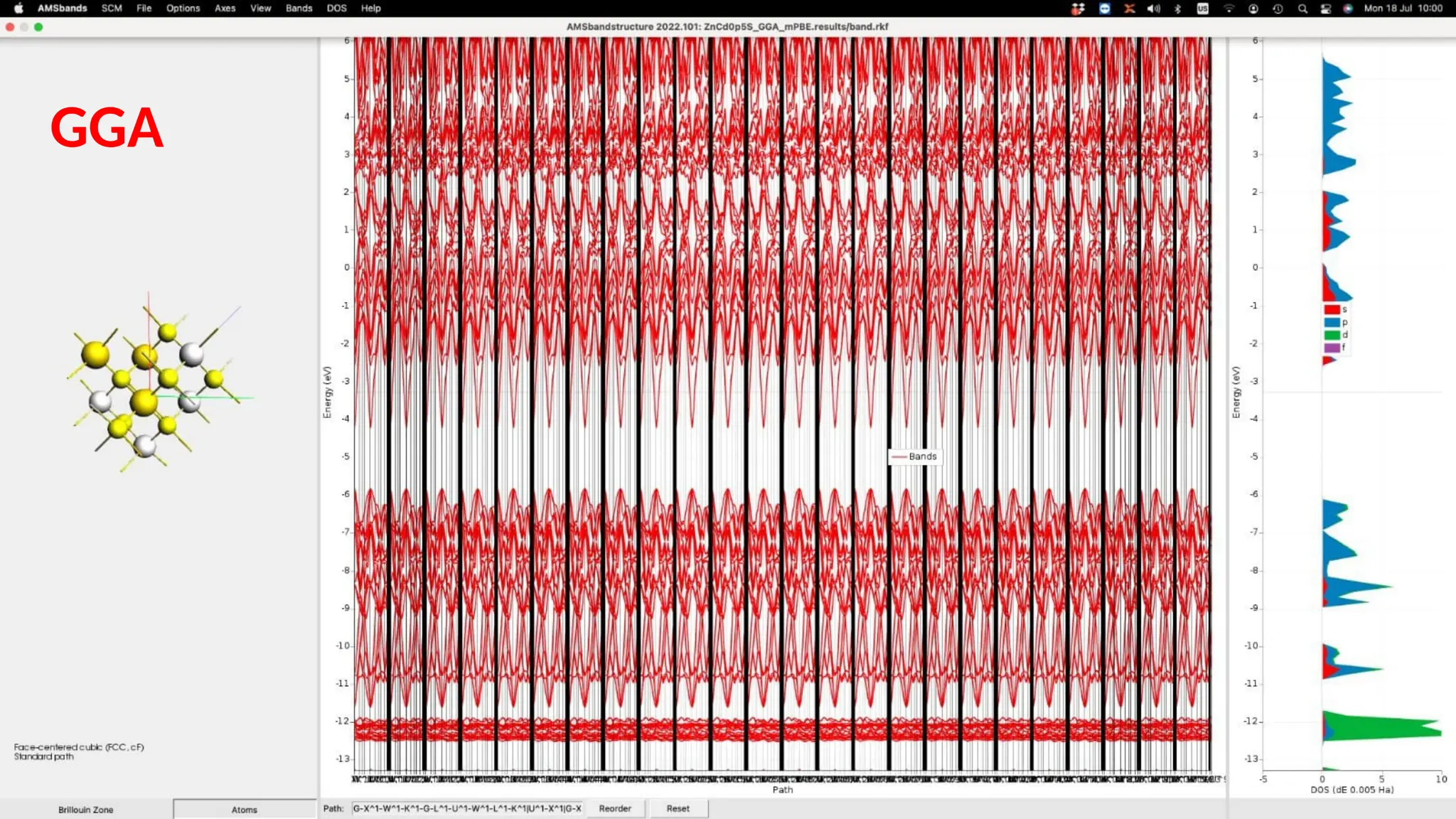
#28 Similarly, GGA-mPBE functional was used to calculate properties of ZnCdS. This is the band structure of 16 atoms used to construct the unit cell. The unit cell volume in this case is again 316.68 angstrom cube. The band gap is also calculated to be 3.64eV as was calculated with LDA. This means that both LDA and GGA-mPBE functionals give similar results in case of ZnCdS thin Structures.
#29 The total density of state plot of GGA-mPBE is shown here in detail. This once again has shown an n-type semiconductor with a large band gap value of 3.64eV which is similar to the LDA approximation.
#30 Here are the partial density of states for Zn s , p and d orbitals and it can be seen that the major contribution is because of the Zn d orbitals.
#31 Here are the partial density of states for Cd s , p and d orbitals and it can be seen that the major contribution is because of the Cd d orbitals.
#32 Here are the partial density of states for S s , p and d orbitals and it can be seen that the major contribution is because of the S p orbitals.