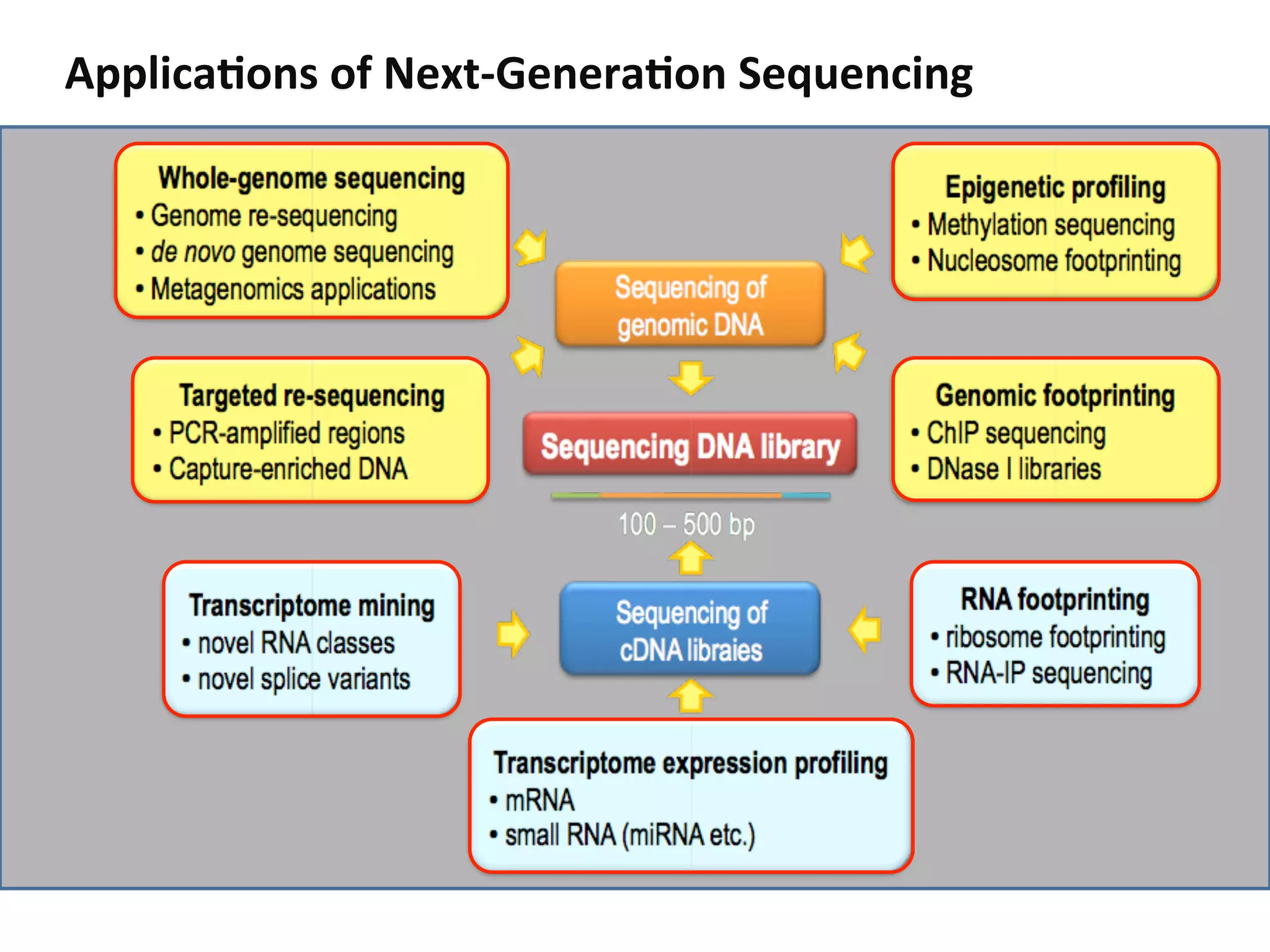
This document provides an introduction to RNA sequencing (RNA-Seq) applications using next-generation sequencing technologies. It discusses how RNA-Seq can be used to identify which genes are expressed, detect differential gene expression between samples, identify splicing isoforms, and detect genetic variants and structural variations. The document reviews Illumina sequencing by synthesis, the most common platform, outlining the work flow from sample acquisition, RNA extraction and library preparation to sequencing. It also discusses considerations for different sample types and extraction methods.
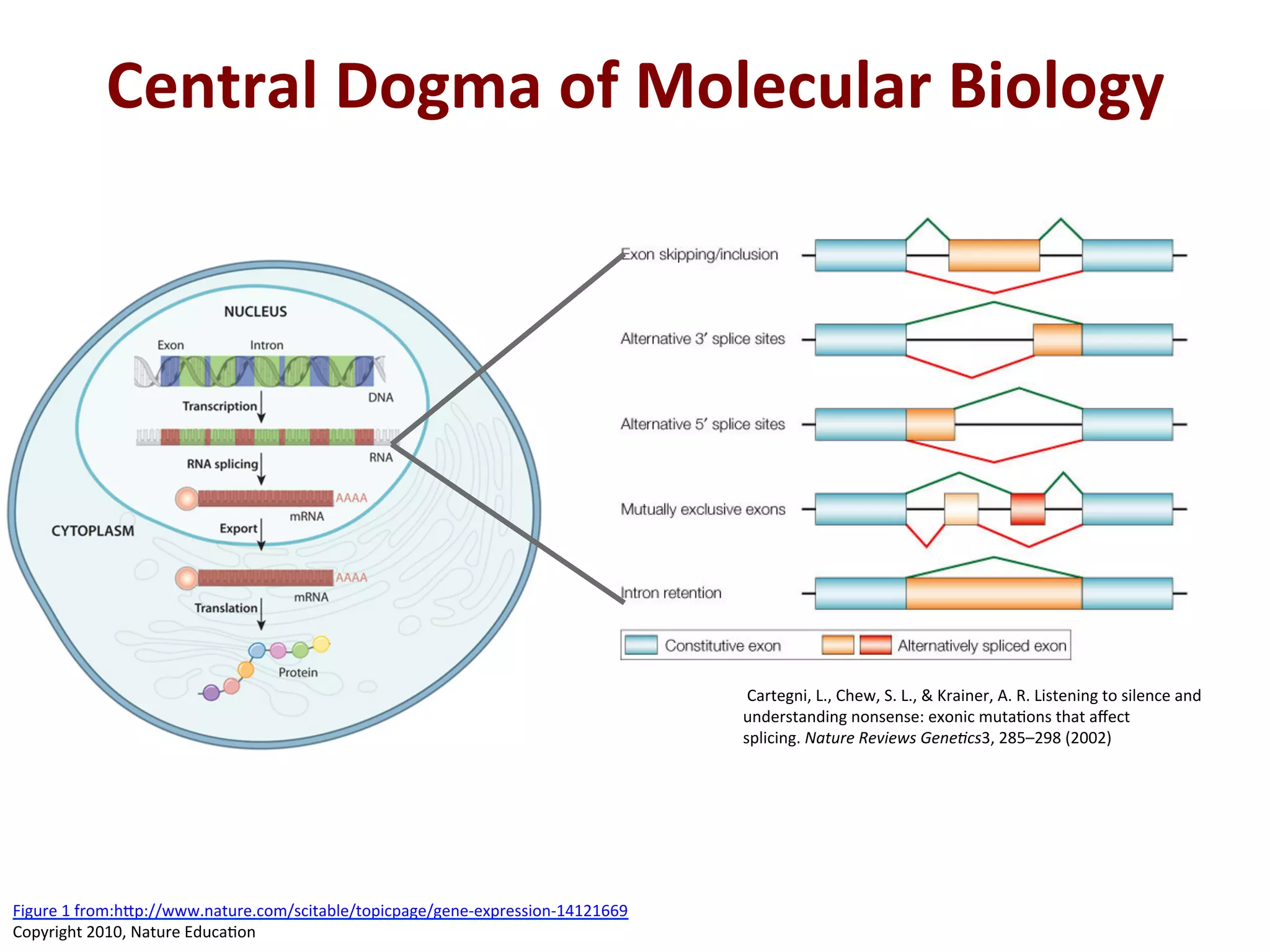
Central
Dogma
of
Molecular
Biology
Cartegni,
L.,
Chew,
S.
L.,
&
Krainer,
A.
R.
Listening
to
silence
and
understanding
nonsense:
exonic
muta%ons
that
affect
splicing.
Nature
Reviews
Gene/cs3,
285–298
(2002)
Figure
1
from:h[p://www.nature.com/scitable/topicpage/gene-‐expression-‐14121669
Copyright
2010,
Nature
Educa%on
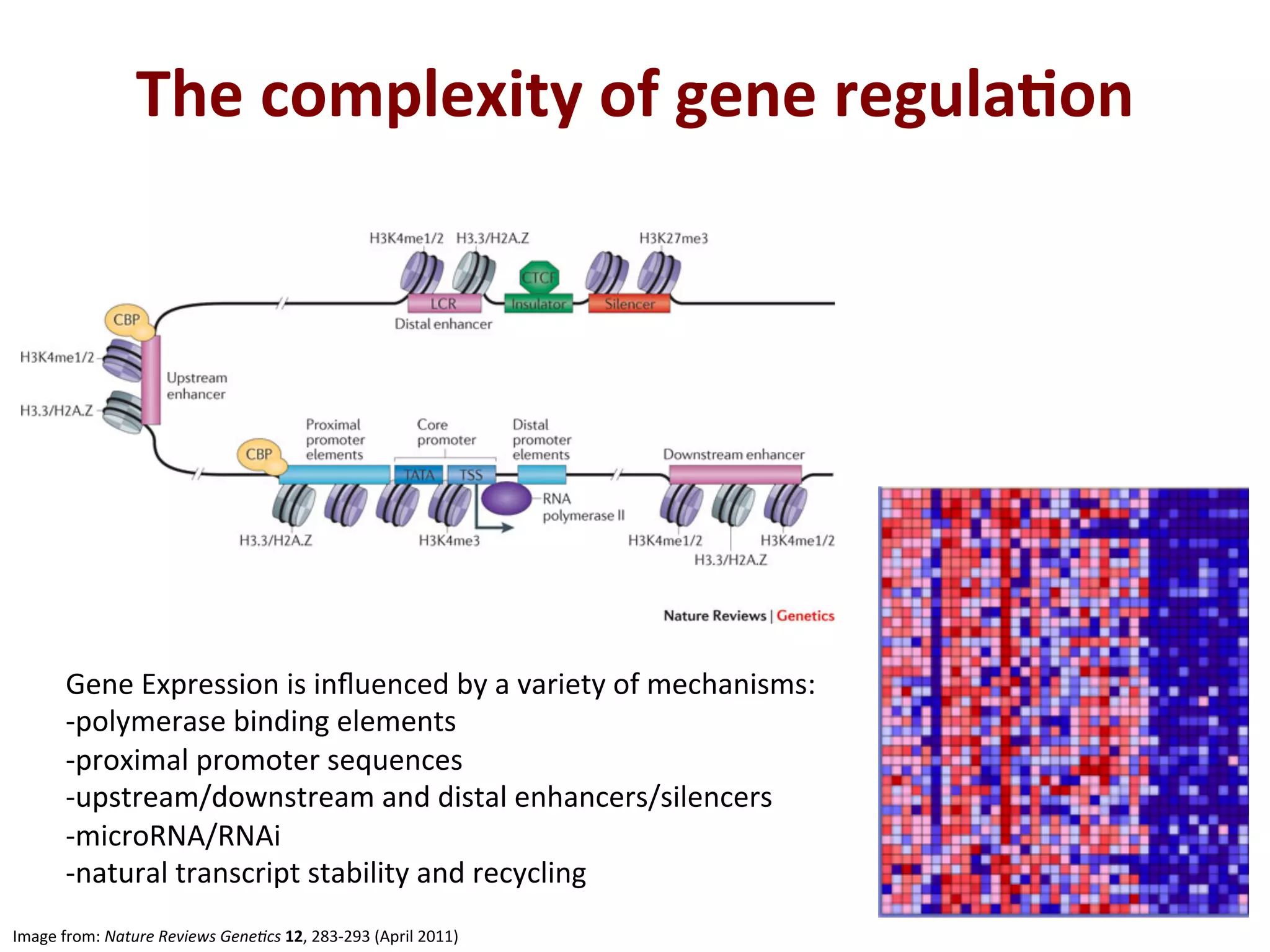
5.
The
complexity
of
gene
regula+on
Image
from:
Nature
Reviews
Gene/cs
12,
283-‐293
(April
2011)
Gene
Expression
is
influenced
by
a
variety
of
mechanisms:
-‐polymerase
binding
elements
-‐proximal
promoter
sequences
-‐upstream/downstream
and
distal
enhancers/silencers
-‐microRNA/RNAi
-‐natural
transcript
stability
and
recycling
6.
What
ques+ons
do
we
want
to
answer?
SNP
and
Indel
Detec%on
REF
ATCGGTACCATCCAGCTAAGGCT
S1
ATCGGAACCATCCAGCTAACGCT
S2
ATCGGTACCATC-‐-‐-‐CTAAGGCT
S3
ATCGGAACCATCCAGCTAAGGCT
S4
ATCGGTA-‐-‐-‐-‐-‐-‐-‐-‐CTAAGGCT
• Which
genes
are
expressed?
• In
experiments
with
mul%ple
samples,
which
genes
exhibit
differen%al
expression?
• Can
we
detect
splicing
isoforms
expression?
• Can
we
detect
novel
genes
or
isoforms?
• Can
we
detect
structural
variants?
SNPs,
inser%ons,
dele%ons,
RNA-‐edi%ng.
• Can
we
detect
ncRNA
that
controls
gene
regula%on
• Can
we
use
differen%al
expression
to
construct
biomarkers
for
diseases?
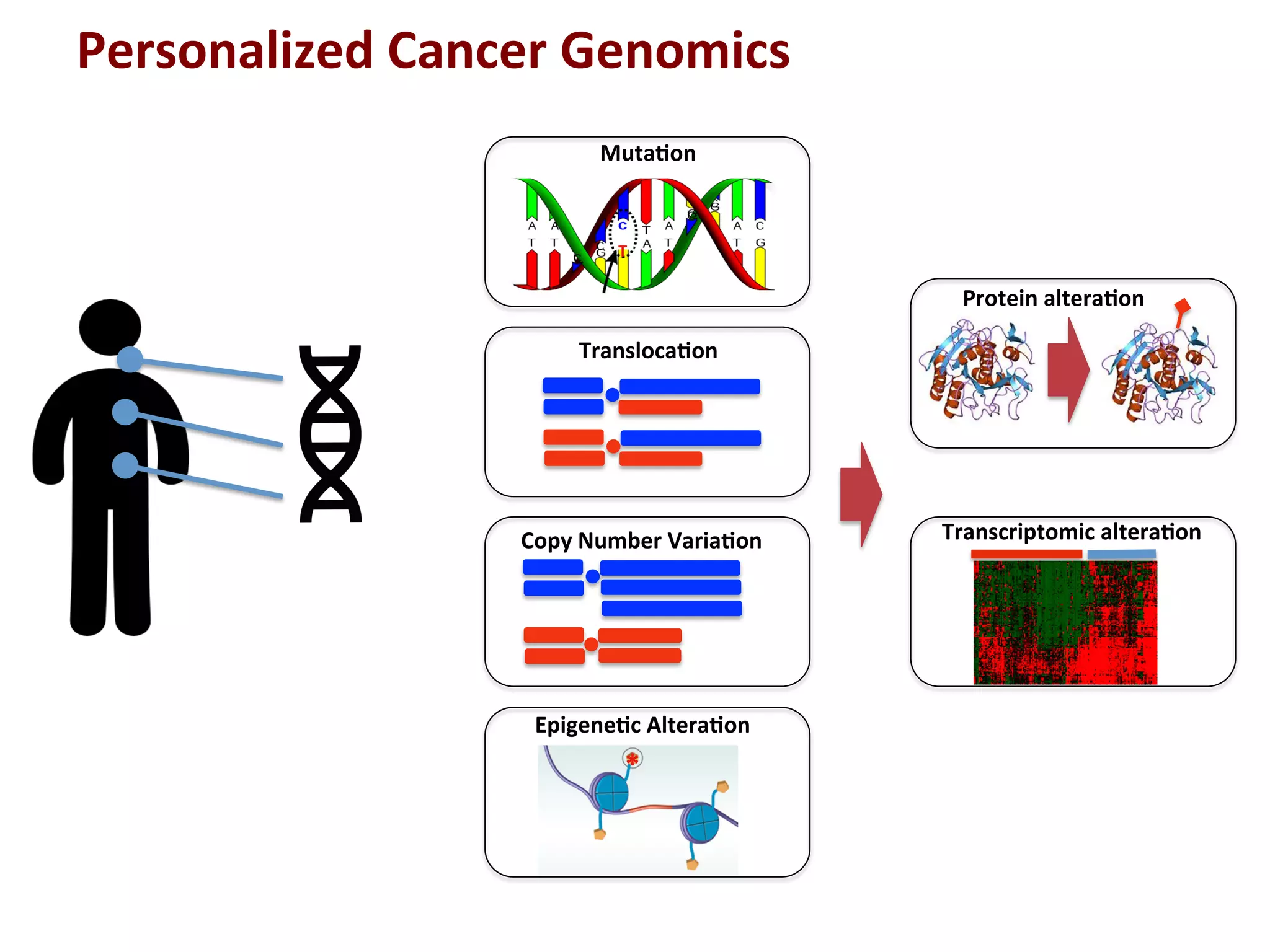
7.
Personalized
Cancer
Genomics
Muta+on
Transloca+on
Copy
Number
Varia+on
Epigene+c
Altera+on
Protein
altera+on
Transcriptomic
altera+on
T
*
8.
What
is
RNASeq?
RNASeq
means
the
sequencing
of
RNA
using
NGS
technology,
which
means
that…..
• Any
type
of
RNA
from
any
sample
sources,
such
as
cell,
body
fluid,
stool,
water,
etc.
can
be
the
sequenced
• Sample
from
different
sample
source
require
different
extrac%on
method
• Different
RNA
species
with
different
sizes
(i.e.
miRNA,
snoRNA,
tRNA)
require
different
prepara%on
protocol
• RNASeq
very
strictly
refers
to
the
sequencing
of
mRNA
from
cells
in
this
course
9.
What
is
RNASeq
Analysis?
• Also
known
as
Whole
Transcriptome
Shotgun
Sequencing
• Iden%fica%on
and
quan%fica%on
of
RNA
snapshot
from
a
genome
at
a
specific
%me
point
• Method
to
study
how
genes
are
being
regulated
for
a
give
cell
type
(i.e.
tumor
cells
v.s.
normal
cells)
at
a
given
%me
using
Next
Genera%on
Sequencing
(NGS)
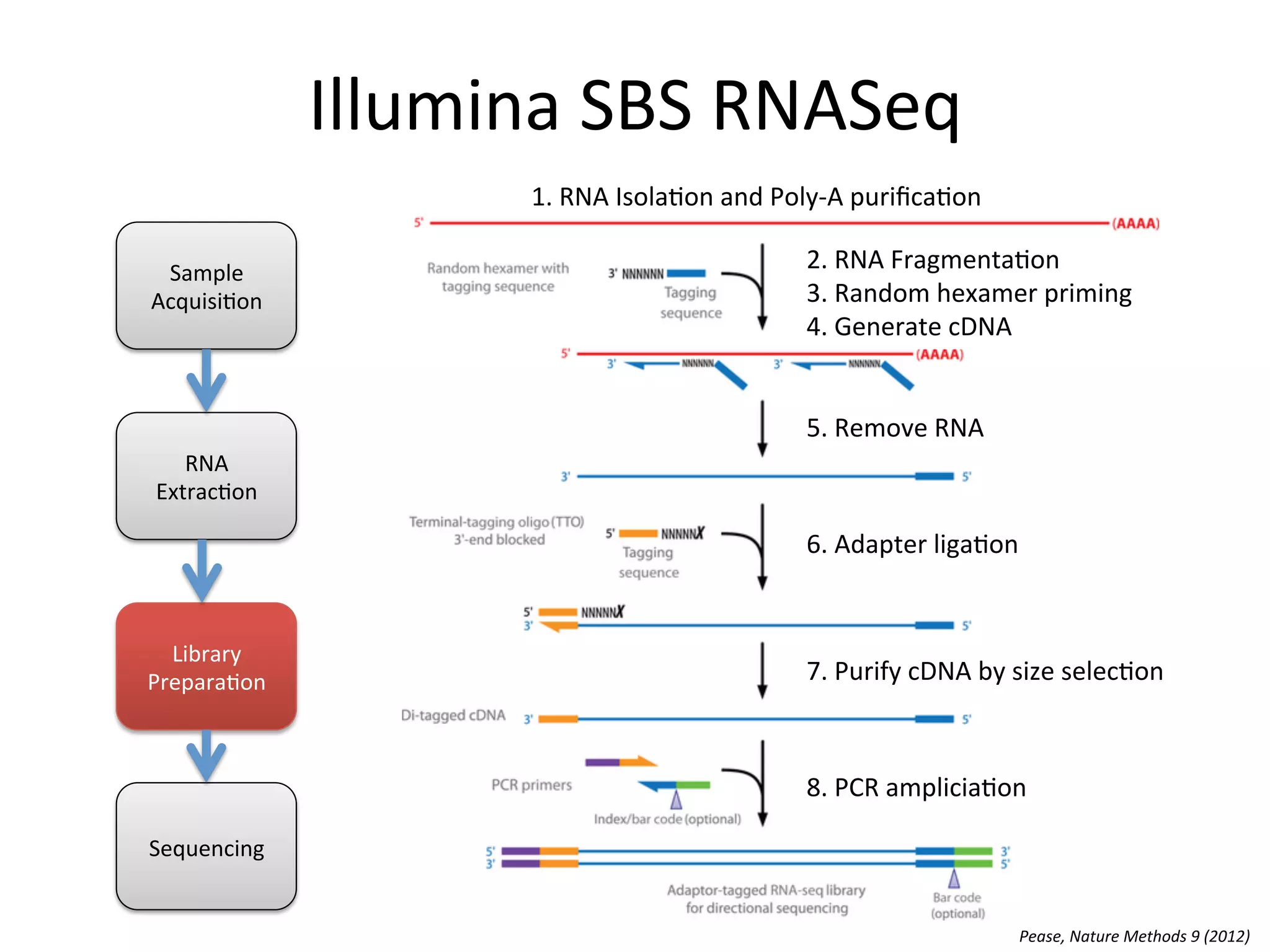
Illumina
SBS
RNASeq
Work
Flow
Sample
Acquisi%on
RNA
Extrac%on
Library
Prepara%on
Sequencing
14.
Illumina
SBS
RNASeq
Work
Flow
Sample
Acquisi%on
RNA
Extrac%on
Library
Prepara%on
Sequencing
Fresh
Frozen
Tissues
-‐ Sample
%ssues
freeze
to
-‐80C
or
immerse
in
liquid
nitrogen
shortly
aler
sample
extrac%on
-‐ All
RNA
is
intact
in
natural
form
but
with
slow
degrada%on
process
-‐ Produce
highest
quality
data
-‐ Expensive
to
keep
and
rare
to
acquire
Formalin
Fixed
Paraffin
Embedded
(FFPE)
Samples
-‐ Fix
sample
%ssues
in
paraffin
wax
immediately
aler
extrac%on
-‐ All
RNA
are
immediately
sheared
into
fragments
-‐ All
mature
mRNA
lost
poly-‐A
tail
-‐ Most
common
sample
available
from
clinic
-‐ Used
in
pathology
lab
-‐ Very
cheap
to
store
15.
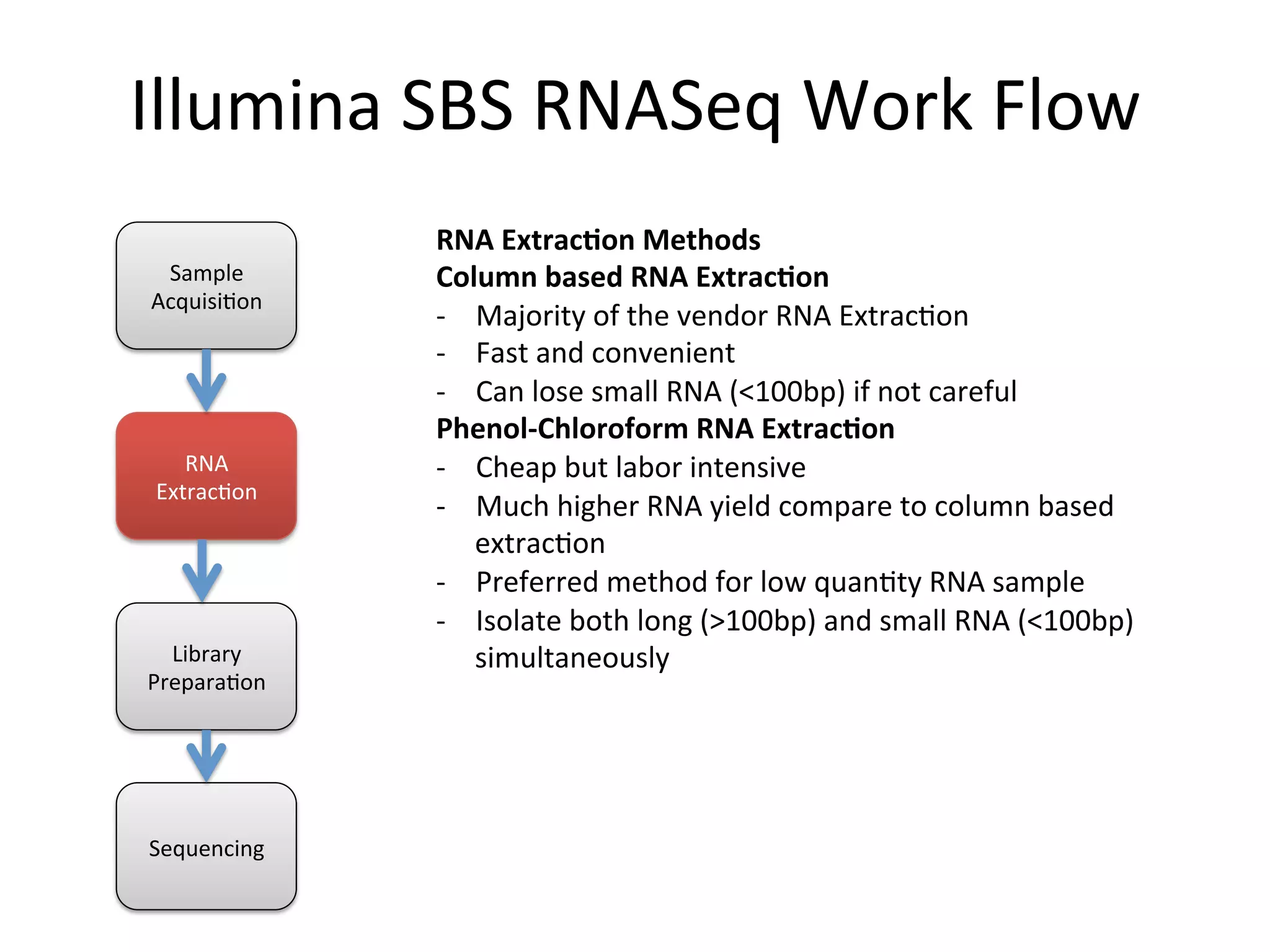
Illumina
SBS
RNASeq
Work
Flow
RNA
Extrac+on
Methods
Column
based
RNA
Extrac+on
-‐ Majority
of
the
vendor
RNA
Extrac%on
-‐ Fast
and
convenient
-‐ Can
lose
small
RNA
(<100bp)
if
not
careful
Phenol-‐Chloroform
RNA
Extrac+on
-‐ Cheap
but
labor
intensive
-‐ Much
higher
RNA
yield
compare
to
column
based
extrac%on
-‐ Preferred
method
for
low
quan%ty
RNA
sample
-‐ Isolate
both
long
(>100bp)
and
small
RNA
(<100bp)
simultaneously
Sample
Acquisi%on
RNA
Extrac%on
Library
Prepara%on
Sequencing
16.
Illumina
SBS
RNASeq
Sample
Acquisi%on
RNA
Extrac%on
Library
Prepara%on
Sequencing
80%
15%
5%
RNA
Composi+on
within
an
eukaryo+c
cell
rRNA
tRNA
Other
RNA
• Pre-‐mRNA
and
mature
mRNA
composed
of
very
small
por%on
of
total
RNA
• MicroRNA,
ncRNA,
and
others
composed
of
even
smaller
number
17.
Illumina
SBS
RNASeq
Library
Prepara%on
Work
Flow
for
mature
mRNA
-‐ RNA
Isola%on
-‐ Poly-‐A
Purifica%on
-‐ Fragmenta%on
-‐ Convert
RNA
to
cDNA
using
random
primers
-‐ Adapter
liga%on
-‐ Size
selec%on
-‐ PCR
amplifica%on
Sample
Acquisi%on
RNA
Extrac%on
Library
Prepara%on
Sequencing
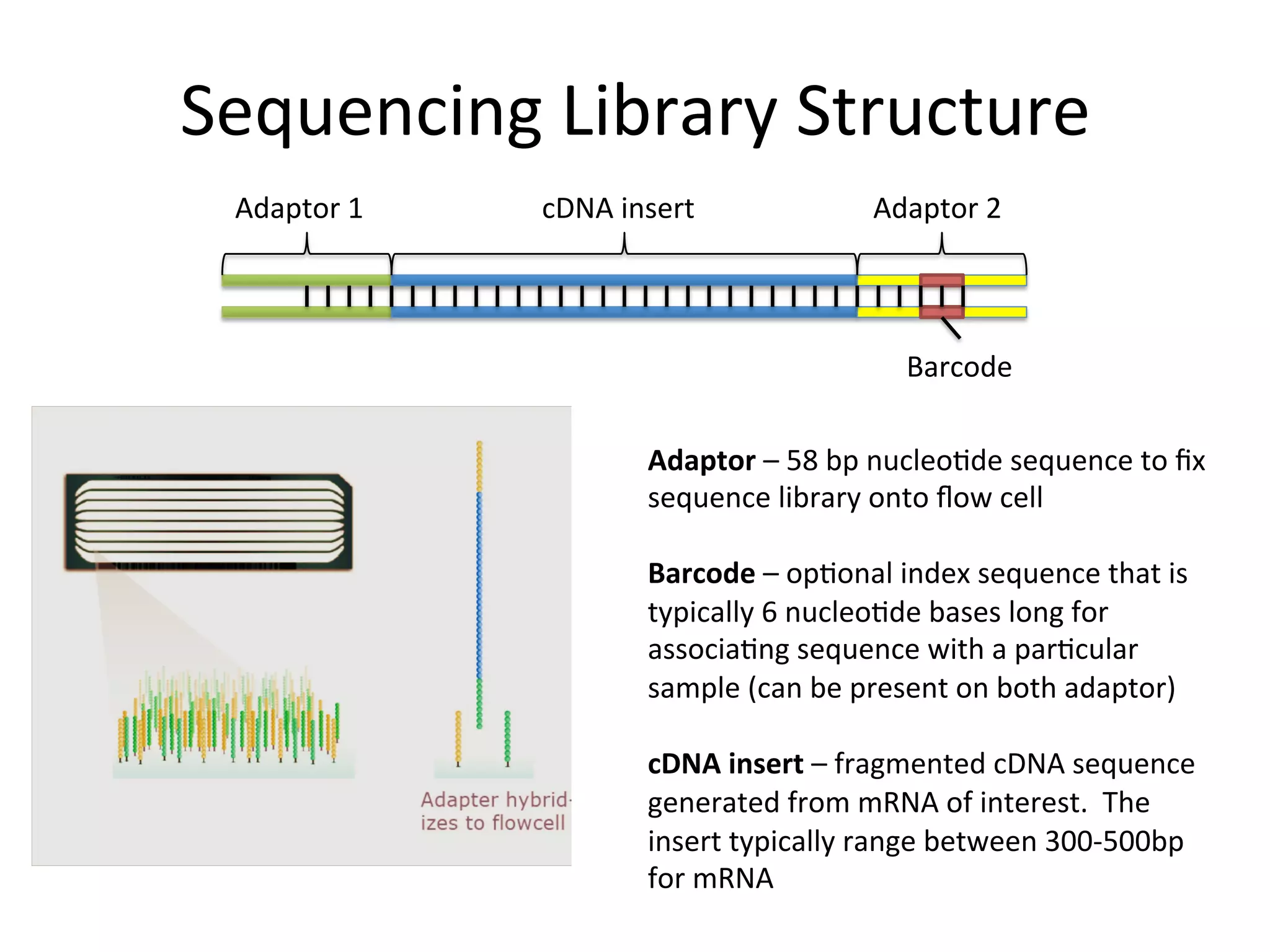
Sequencing
Library
Structure
Adaptor
1
cDNA
insert
Adaptor
2
Barcode
Adaptor
–
58
bp
nucleo%de
sequence
to
fix
sequence
library
onto
flow
cell
Barcode
–
op%onal
index
sequence
that
is
typically
6
nucleo%de
bases
long
for
associa%ng
sequence
with
a
par%cular
sample
(can
be
present
on
both
adaptor)
cDNA
insert
–
fragmented
cDNA
sequence
generated
from
mRNA
of
interest.
The
insert
typically
range
between
300-‐500bp
for
mRNA
20.
Illumina
SBS
RNASeq
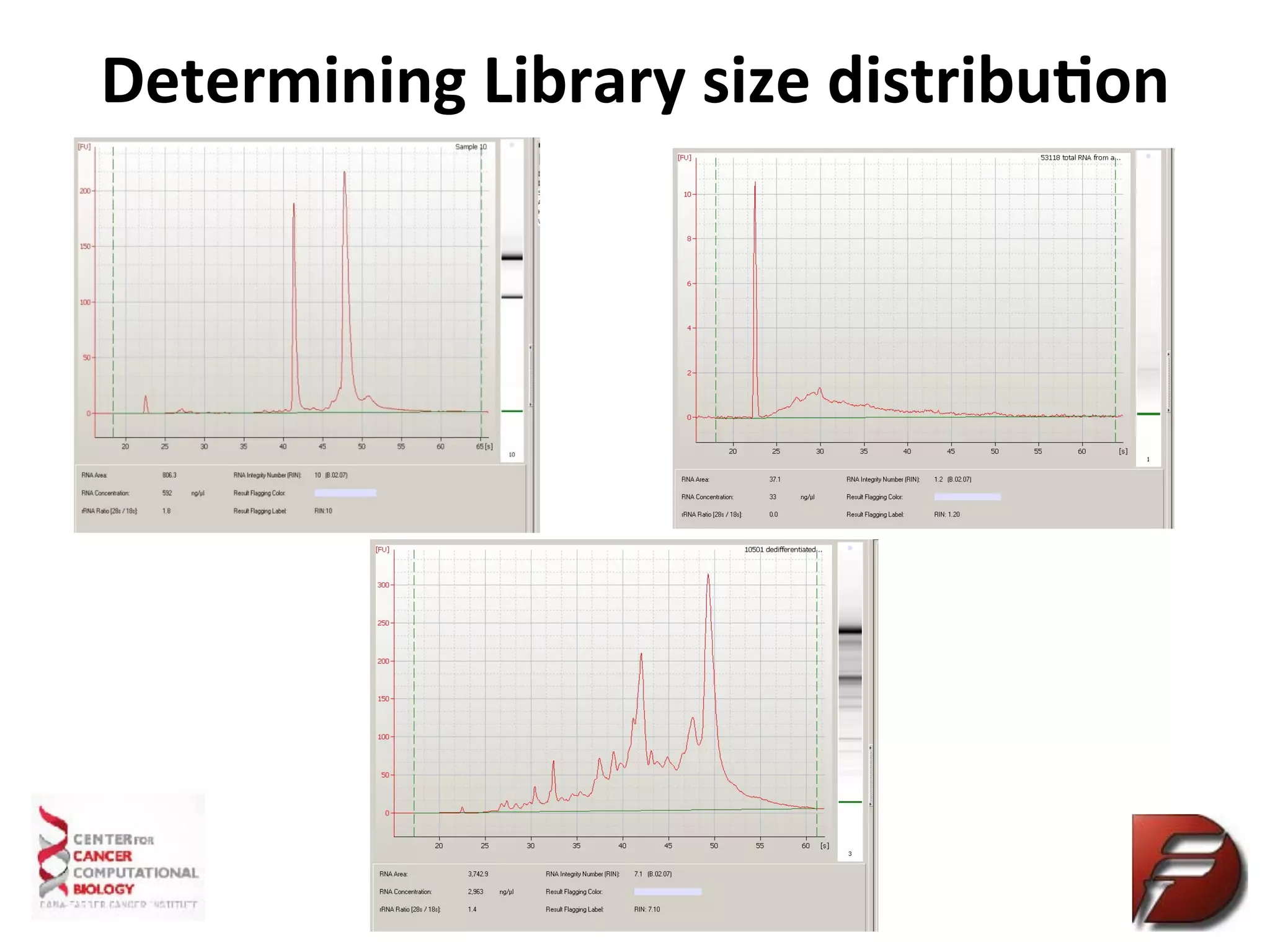
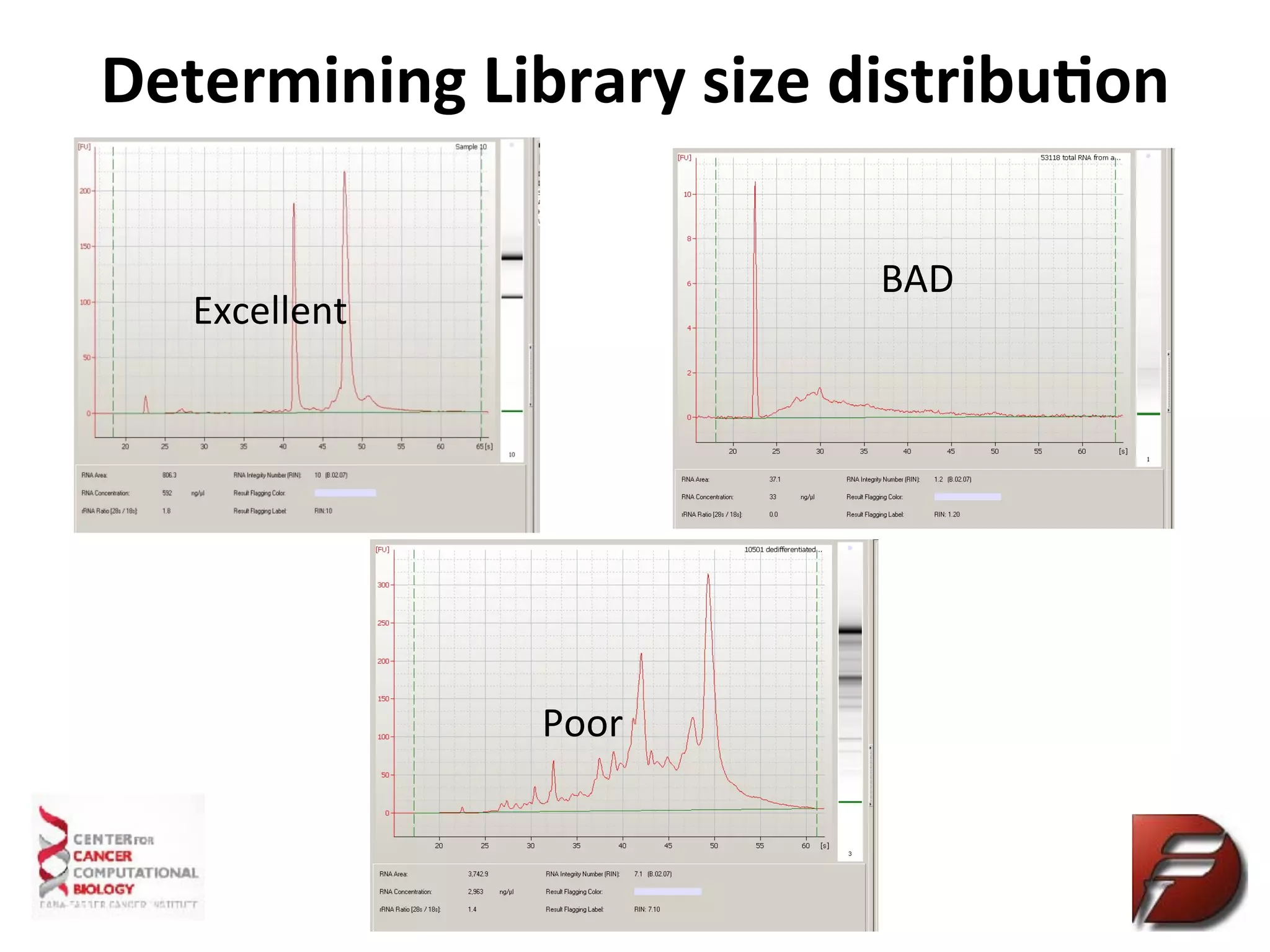
Determine
Sequencing
Library
Quality
Qubit
(RNA)
Measures
the
concentra%on
of
only
double
stranded
DNA,
more
accurate
than
Nanodrop
Bioanalyzer
Measures
the
RNA/library
size
in
base
pairs
qPCR
Measure
the
concentra%on
of
library
that
has
adaptors
ligated
and
will
hybridize
and
sequence
Sample
Acquisi%on
RNA
Extrac%on
Library
Prepara%on
Sequencing
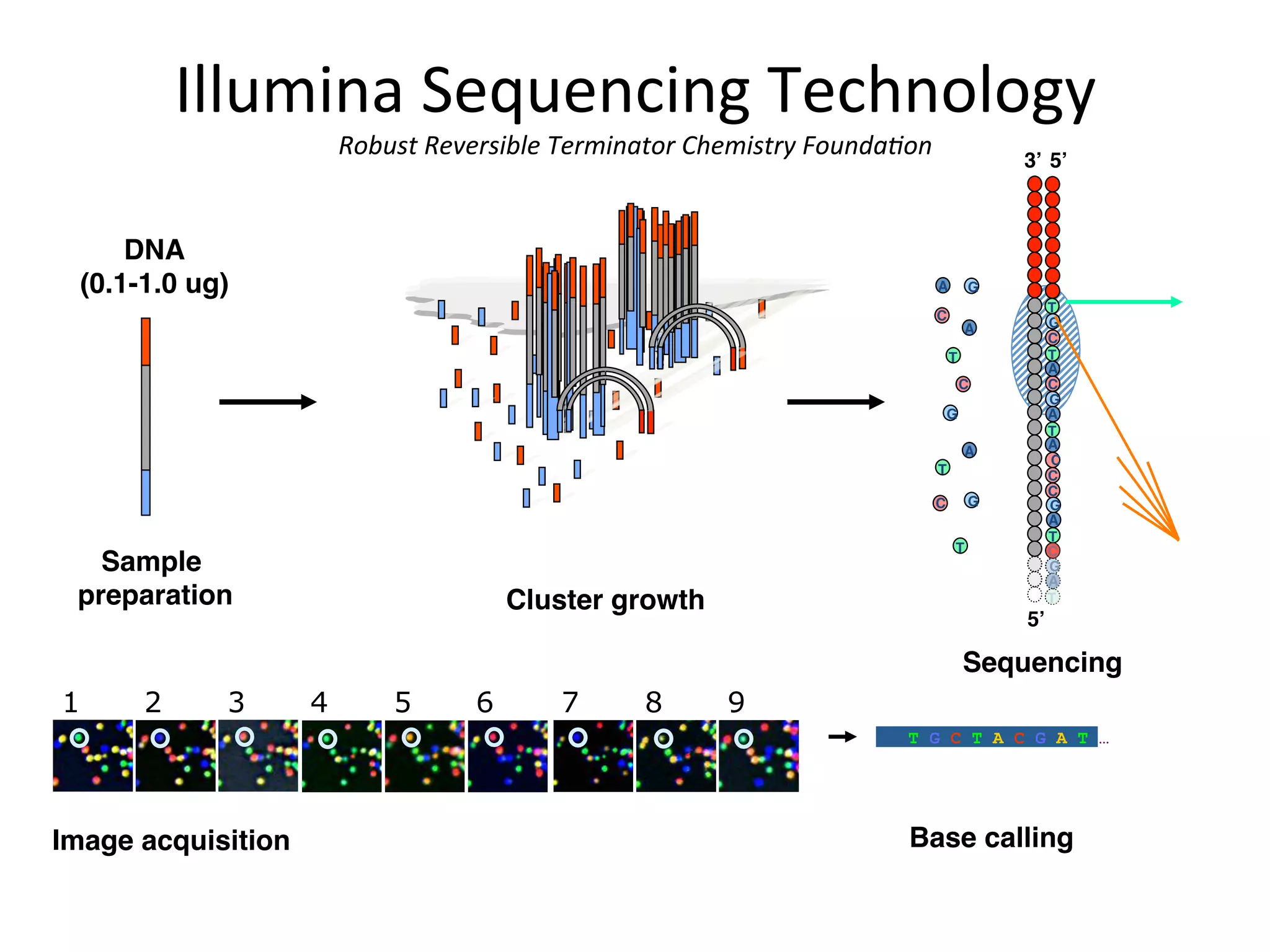
DNA
(0.1-1.0 ug)
"
Single moleculearray"
Sample
preparation" Cluster growth"
5’"
5’"3’"
G"
T"
C"
A"
G"
T"
C"
A"
G"
T"
C"
A"
C"
A"
G"
T"
C"
A"
T"
C"
A"
C"
C"
T"
A"
G"
C"
G"
T"
A"
G"
T"
1 2 3 7 8 94 5 6
Image acquisition" Base calling"
T G C T A C G A T …
Sequencing"
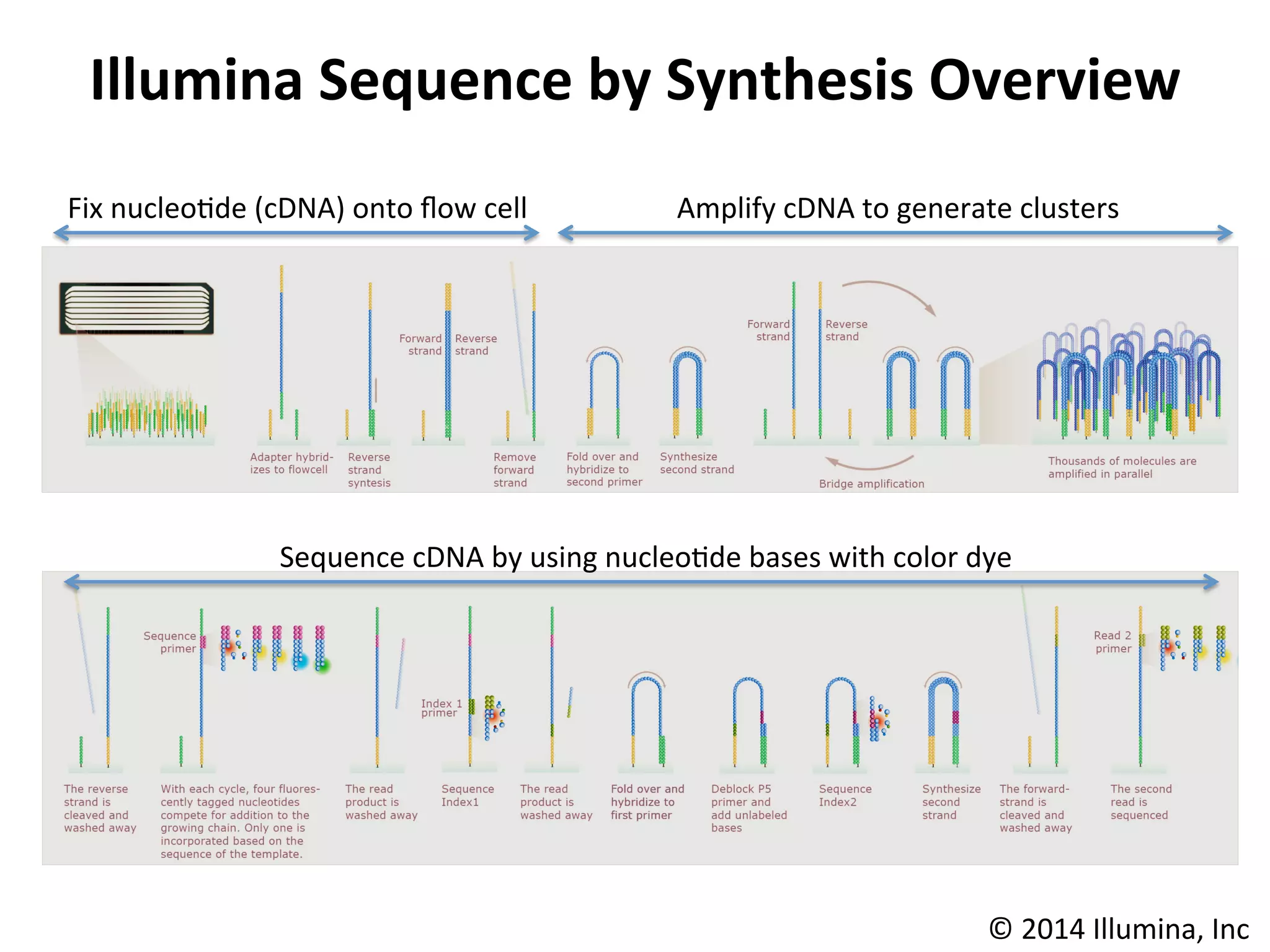
Illumina
Sequencing
Technology
Robust
Reversible
Terminator
Chemistry
Founda/on
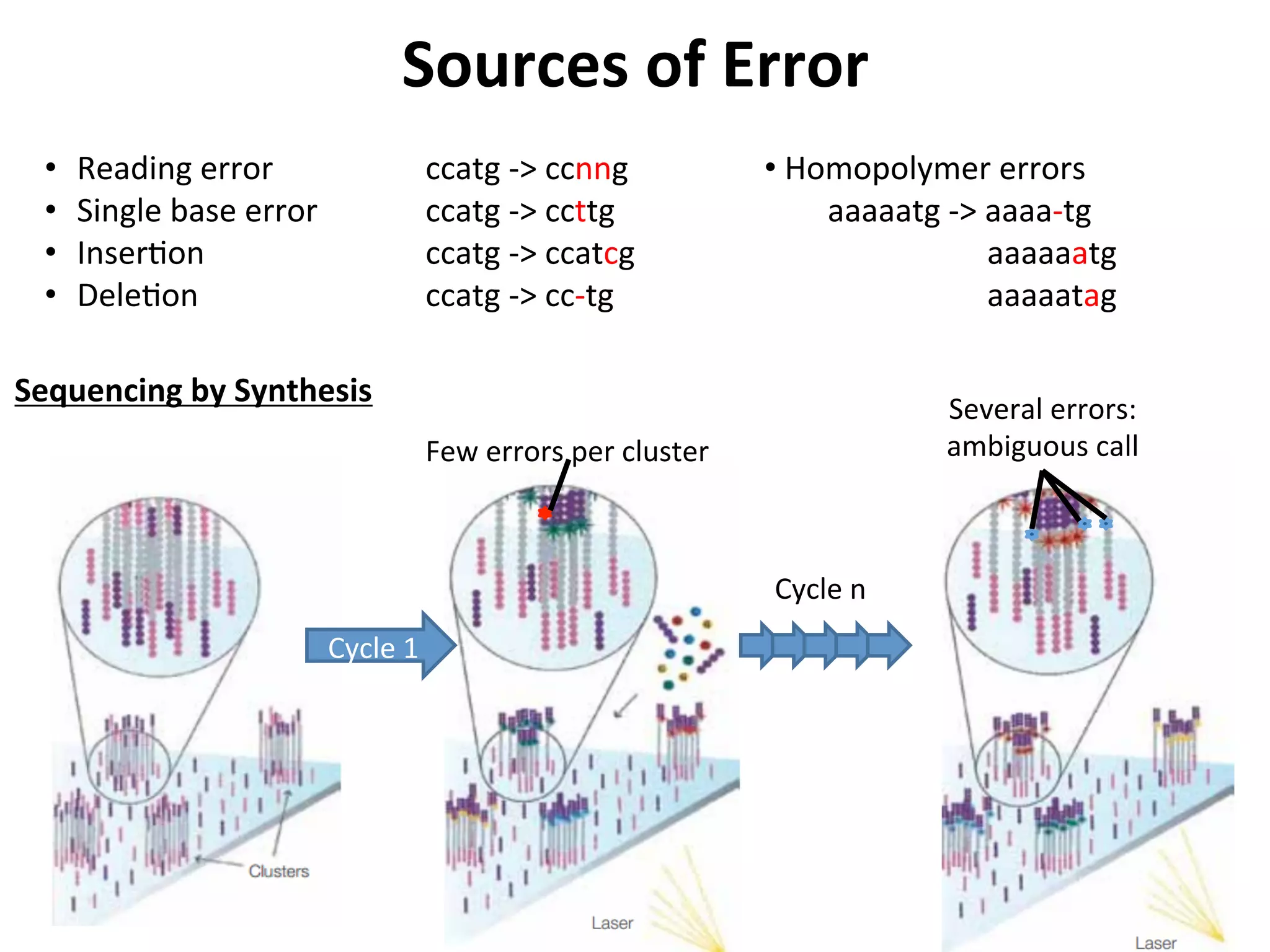
24.
Sources
of
Error
• Reading
error
ccatg
-‐>
ccnng
• Single
base
error
ccatg
-‐>
ccttg
• Inser%on
ccatg
-‐>
ccatcg
• Dele%on
ccatg
-‐>
cc-‐tg
•
Homopolymer
errors
aaaaatg
-‐>
aaaa-‐tg
aaaaaatg
aaaaatag
Cycle
1
Cycle
n
Few
errors
per
cluster
Several
errors:
ambiguous
call
Sequencing
by
Synthesis
25.
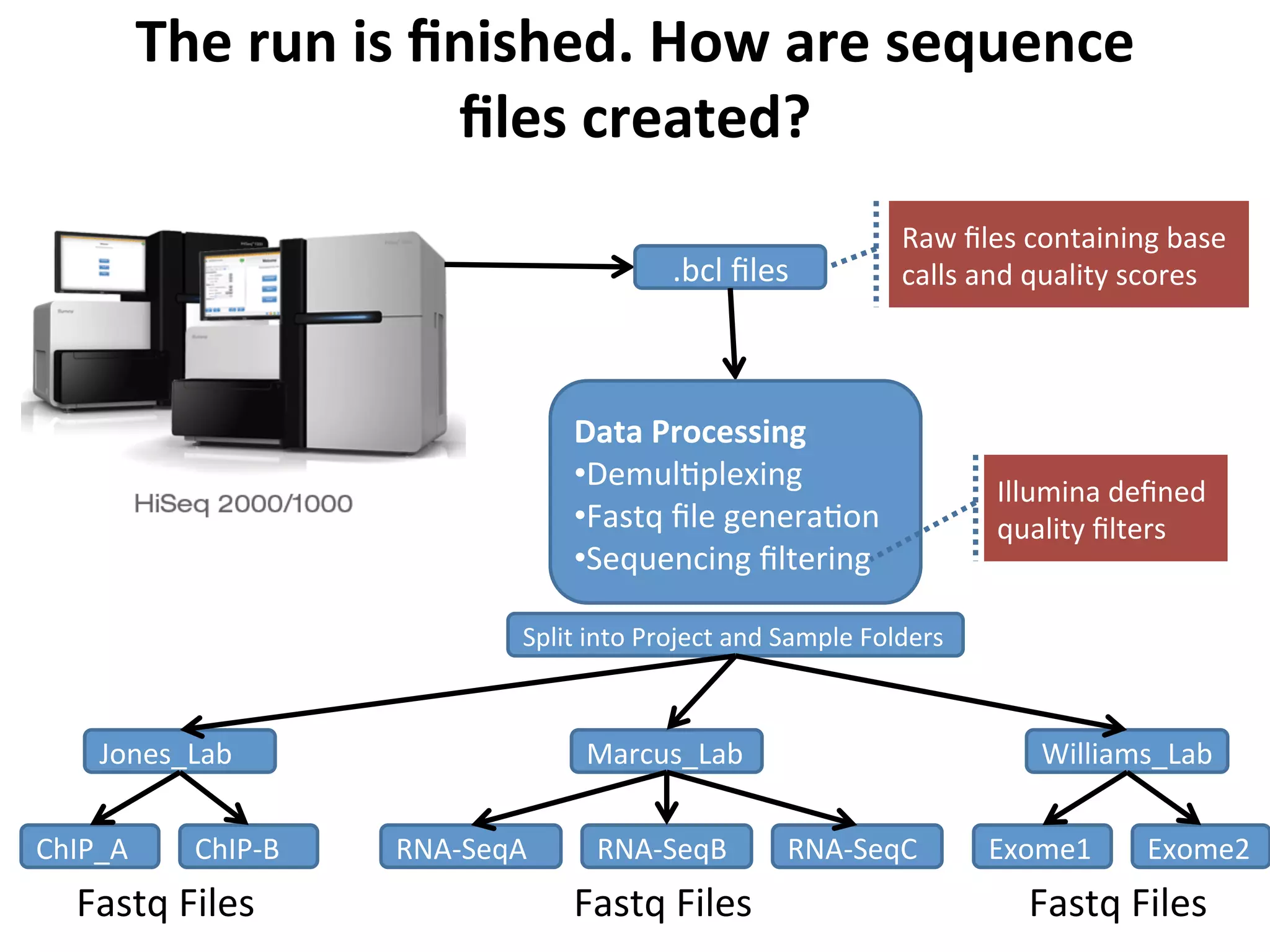
The
run
is
finished.
How
are
sequence
files
created?
.bcl
files
Data
Processing
• Demul%plexing
• Fastq
file
genera%on
• Sequencing
filtering
Raw
files
containing
base
calls
and
quality
scores
Illumina
defined
quality
filters
Split
into
Project
and
Sample
Folders
Jones_Lab
ChIP_A
ChIP-‐B
Marcus_Lab
RNA-‐SeqA
RNA-‐SeqB
RNA-‐SeqC
Williams_Lab
Exome1
Exome2
Fastq
Files
Fastq
Files
Fastq
Files
26.
Illumina
Fastq
Format
Fasta
format
>seqID
CTTCAGACGAGTCGAGGAAAGGCTTTGCTGCTTTCCTTTACAGGGTGGGG
Fastq
format
@HWI-‐ST389:225:D18R8ACXX:5:1101:1421:2191
1:N:0:CCGTCC
CTTCAGACGAGTCGAGGAAAGGCTTTGCTGCTTTCCTTTACAGGGTGGGG
+
@@@DDDDFHHFCFFHIJIHIJGIFGIIHIIIJGIIJHIIJIIJIHDFHJE
Illumina
Fastq
header:
@<instrument>:<run
number>:<flowcell
ID>:<lane>:<%le>:<xpos>:<y-‐
pos><read>:<isfiltered>:<control
number>:<indexsequence>
27.
Illumina
Fastq
Format
Quality
Scores
@@@DDDDFHHFCFFHIJIHIJGIFGIIHIIIJGIIJHIIJIIJIHDFHJE
Illumina
Fastq
header:
@<instrument>:<run
number>:<flowcell
ID>:<lane>:<%le>:<xpos>:<y-‐
pos><read>:<isfiltered>:<control
number>:<indexsequence>
• Each
nucleo%de
in
a
read
has
an
associated
quality
value
(1-‐40).
• The
numerical
value
is
encoded
as
an
ASCII
character
to
save
space.
• Each
q-‐value
represents
a
probability
that
the
nucleo%de
is
incorrect
at
that
posi%on:
Q(X)
=-‐10
log10(P(~X))
Quality
score
Q(A)
Error
probability
P(~A)
10
0.1
20
0.01
30
0.001
40
0.0001
Typical
cutoff
for
acceptable
quality
28.
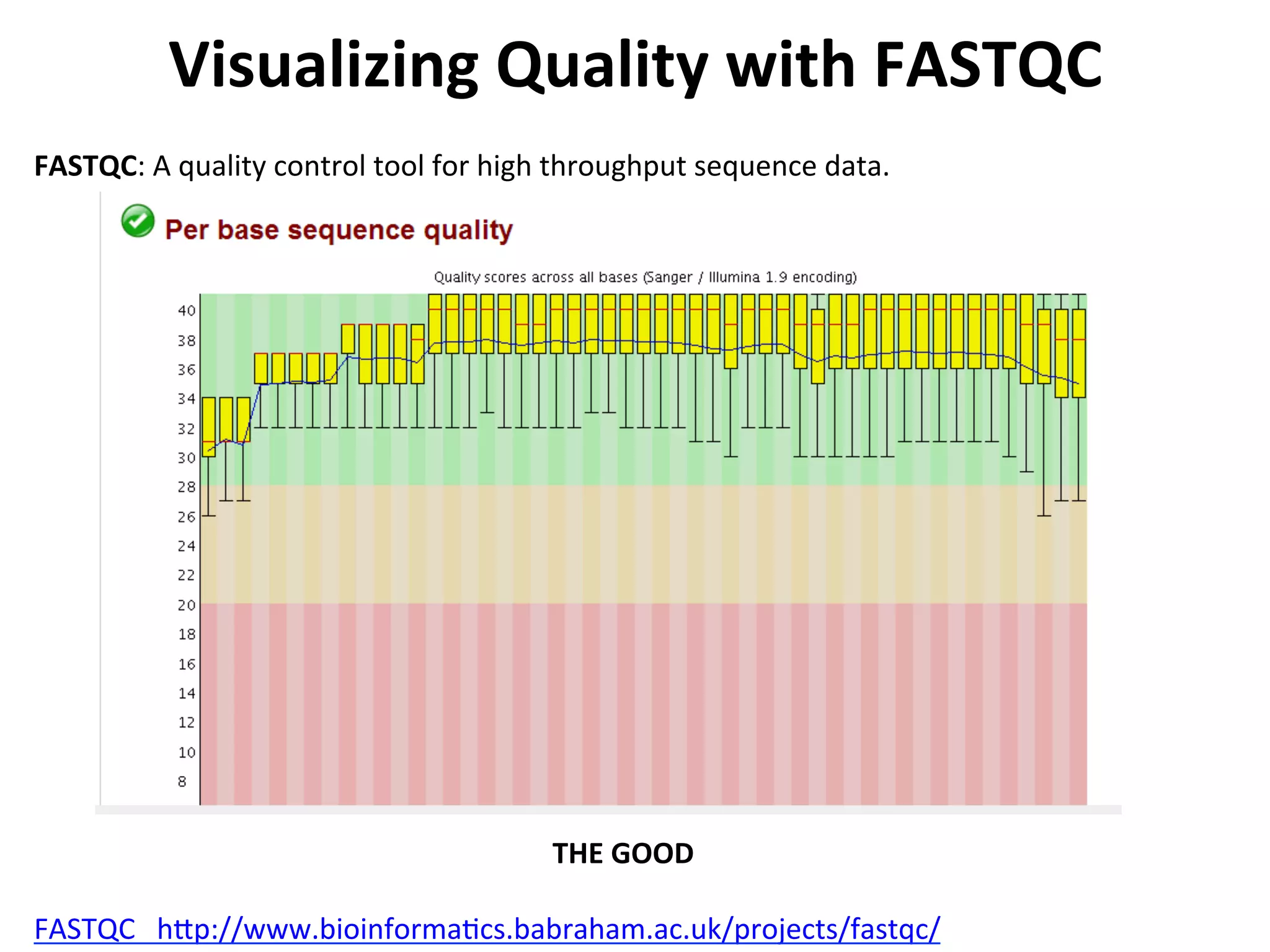
Visualizing
Quality
with
FASTQC
FASTQC
h[p://www.bioinforma%cs.babraham.ac.uk/projects/fastqc/
FASTQC:
A
quality
control
tool
for
high
throughput
sequence
data.
THE
GOOD
29.
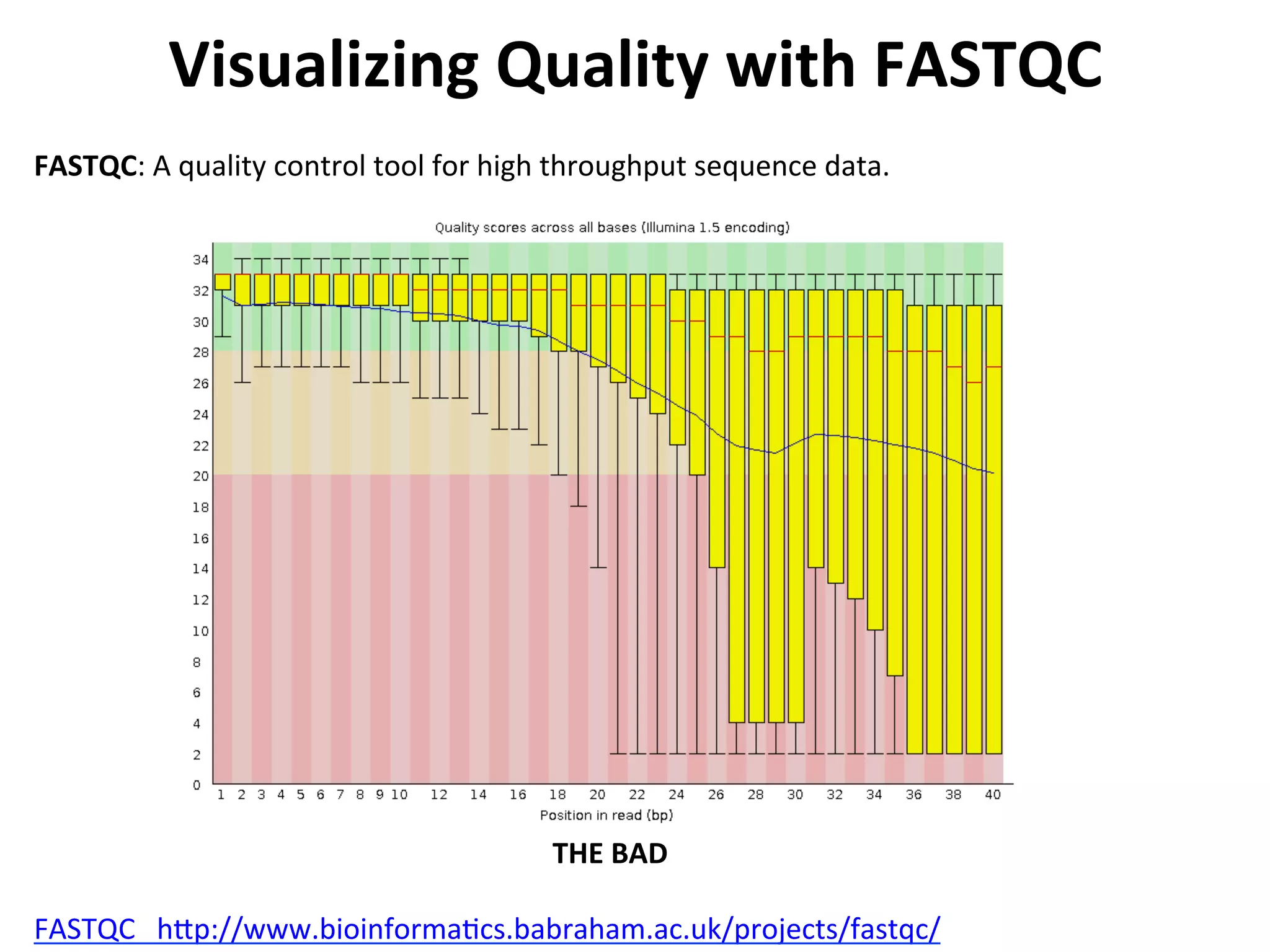
Visualizing
Quality
with
FASTQC
FASTQC
h[p://www.bioinforma%cs.babraham.ac.uk/projects/fastqc/
FASTQC:
A
quality
control
tool
for
high
throughput
sequence
data.
THE
BAD
30.
Data
Quality
Assessment
• Evaluate
read
library
quality
– Determine
if
the
data
is
proper
generated
• No
informa%on
on
if
the
data
is
what
you
want
• Iden%fy
technical
ar%fact
• Iden%fy
poor
quality
samples
• Key
features
to
evaluate
– Uniformity
of
sequencing
quality
score
(phred
score)
– GC
content
distribu%on
– Level
of
sequencing
adapter
contamina%on
– Level
of
sequence
duplica%on
(may
caused
by
PCR
ar%fact,
rRNA
contamina%on,
bacterial
contamina%on)
• Filter
or
trim
data
as
needed
using
FASTX
31.
Use
FASTQC
on
GALAXY
FASTQC
-‐
provide
a
simple
way
to
do
some
quality
control
checks
on
raw
sequence
data
coming
from
high
throughput
sequencing
pipelines.
(h[p://www.bioinforma%cs.babraham.ac.uk/projects/fastqc/)
GALAXY-‐
a
scien%fic
workflow,
data
integra%on,and
data
and
analysis
persistence
and
publishing
plaform
that
aims
to
make
computa%onal
biology
accessible
to
research
scien%sts
that
do
not
have
computer
programming
experience.
(h[ps://galaxyproject.org/)