Downloaded 102 times


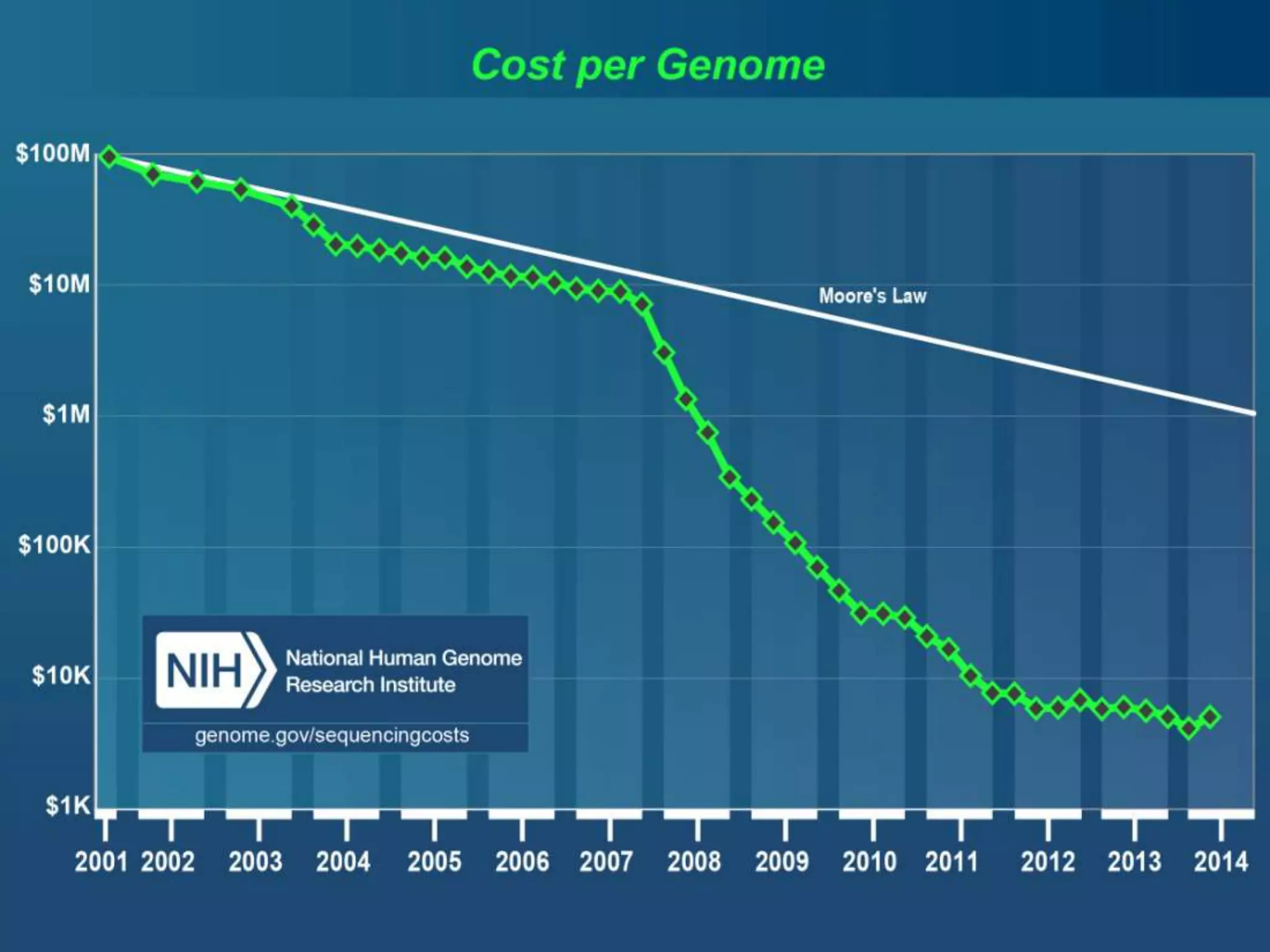
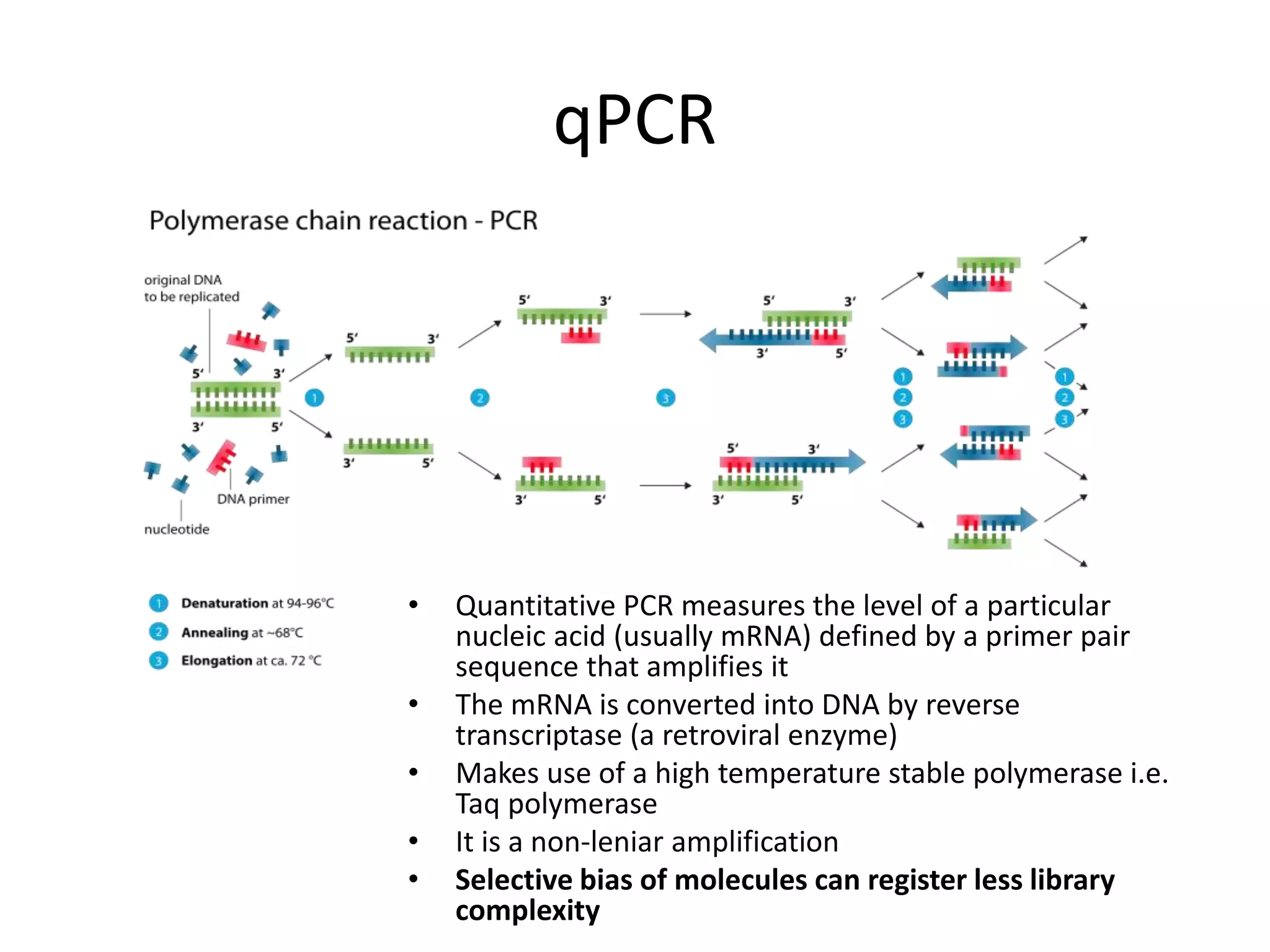

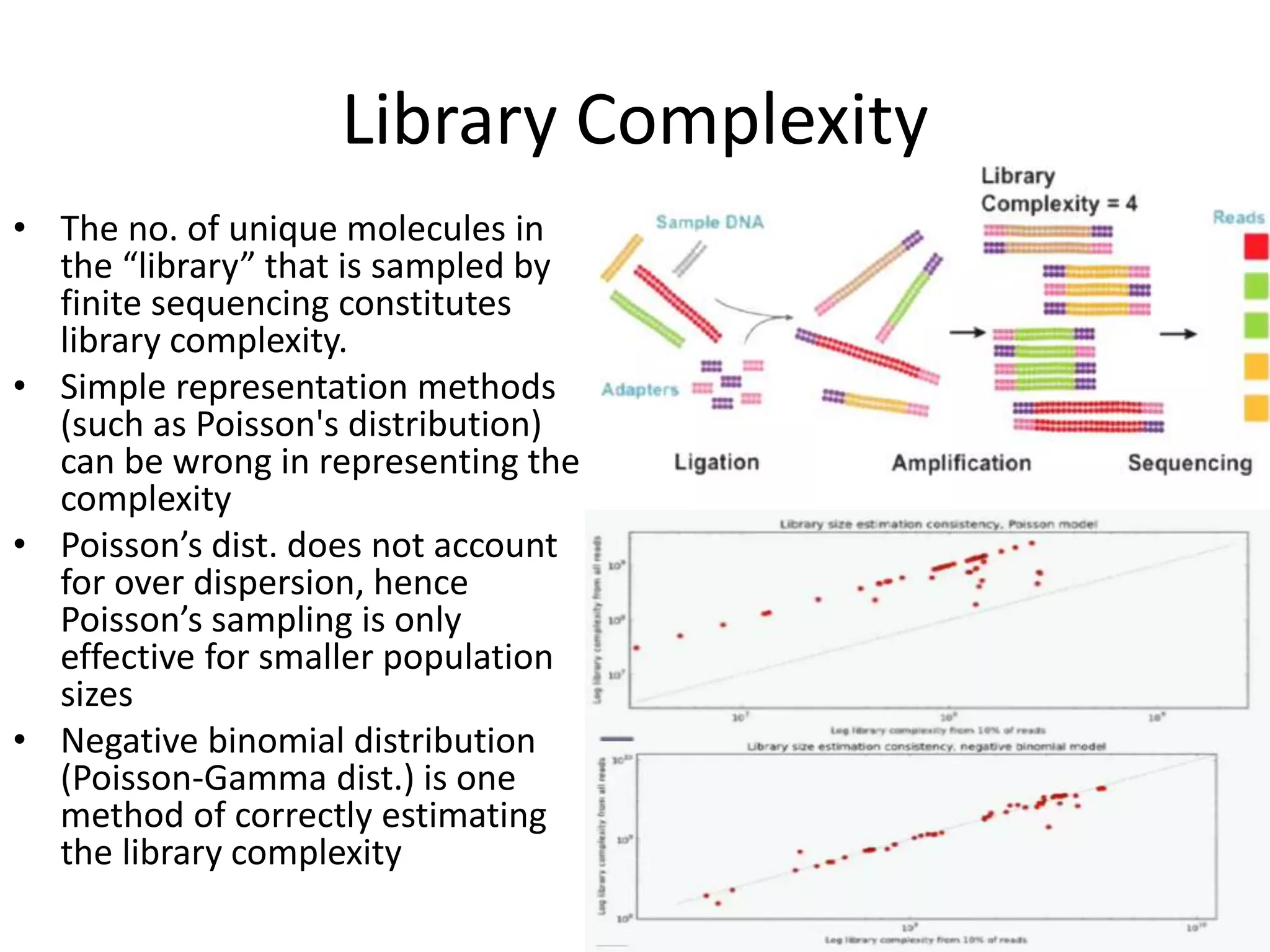

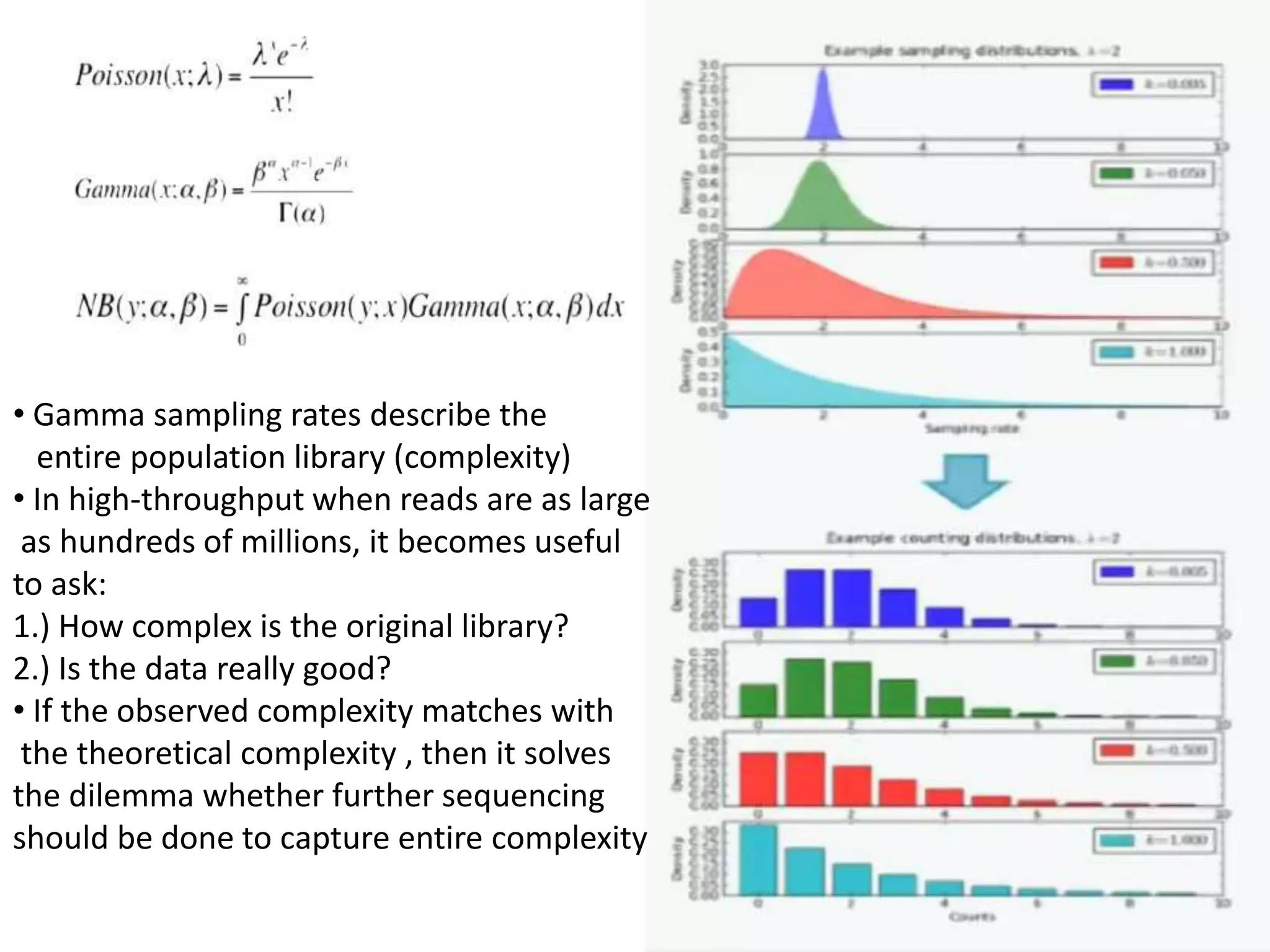

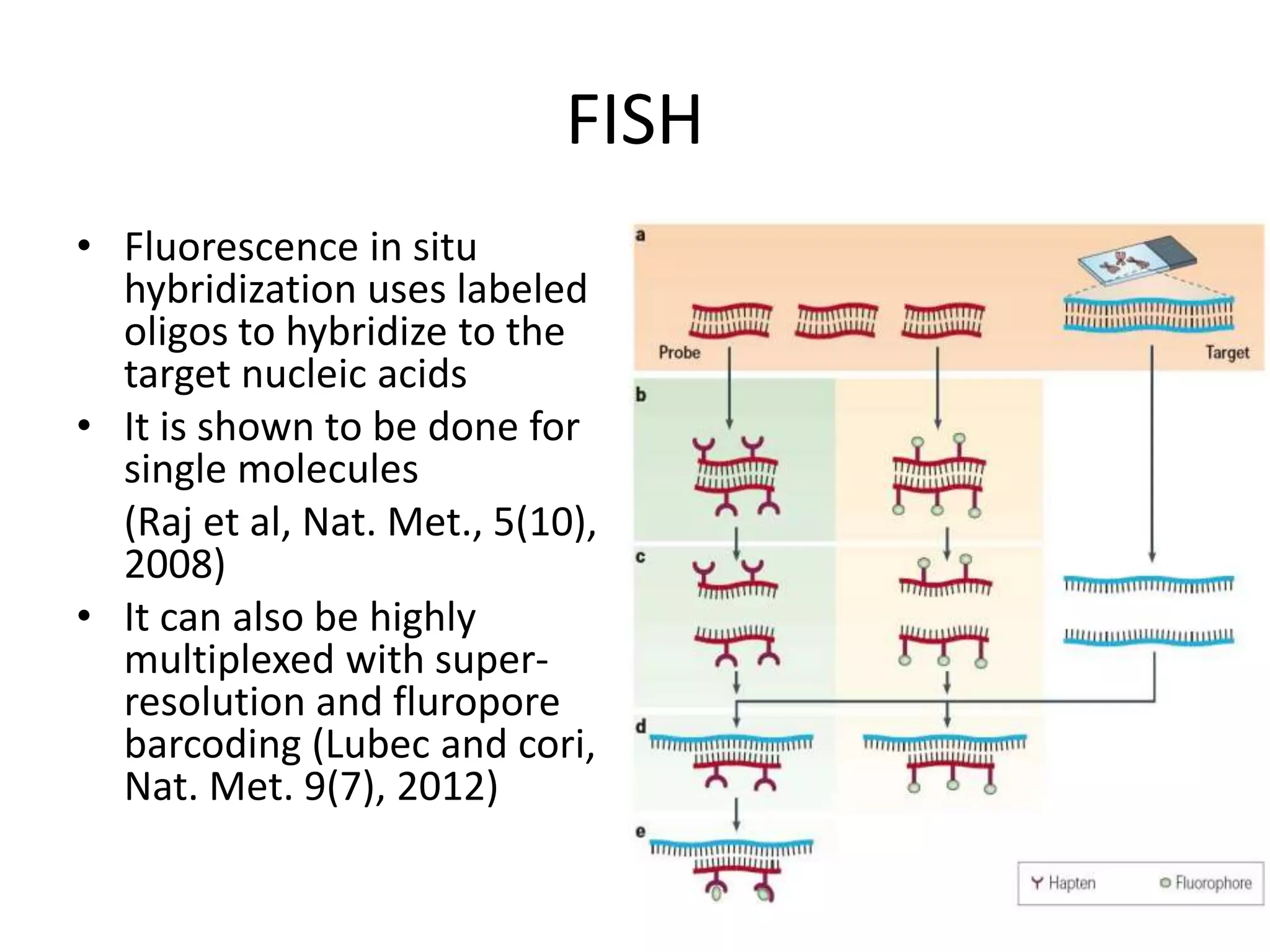


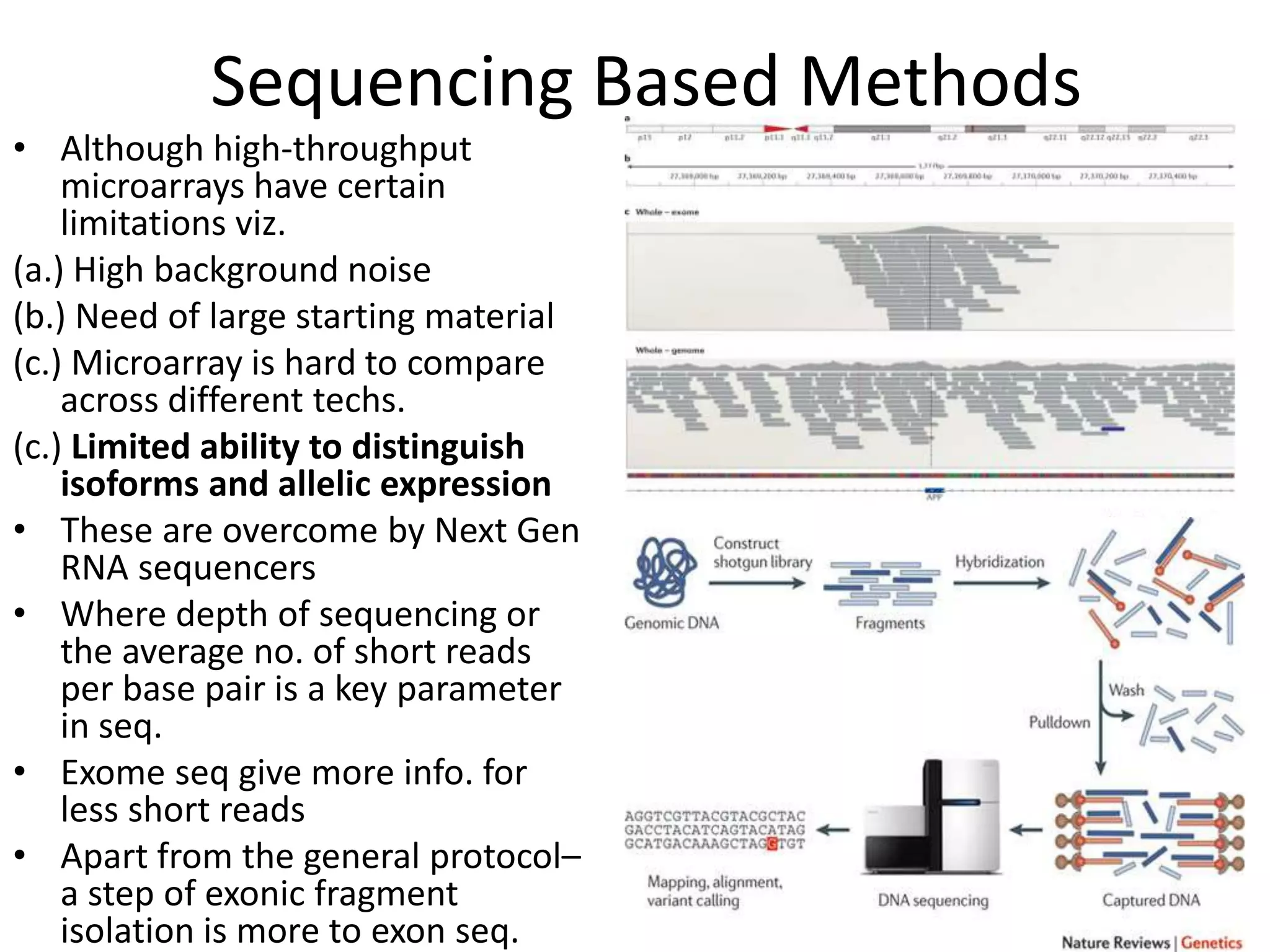
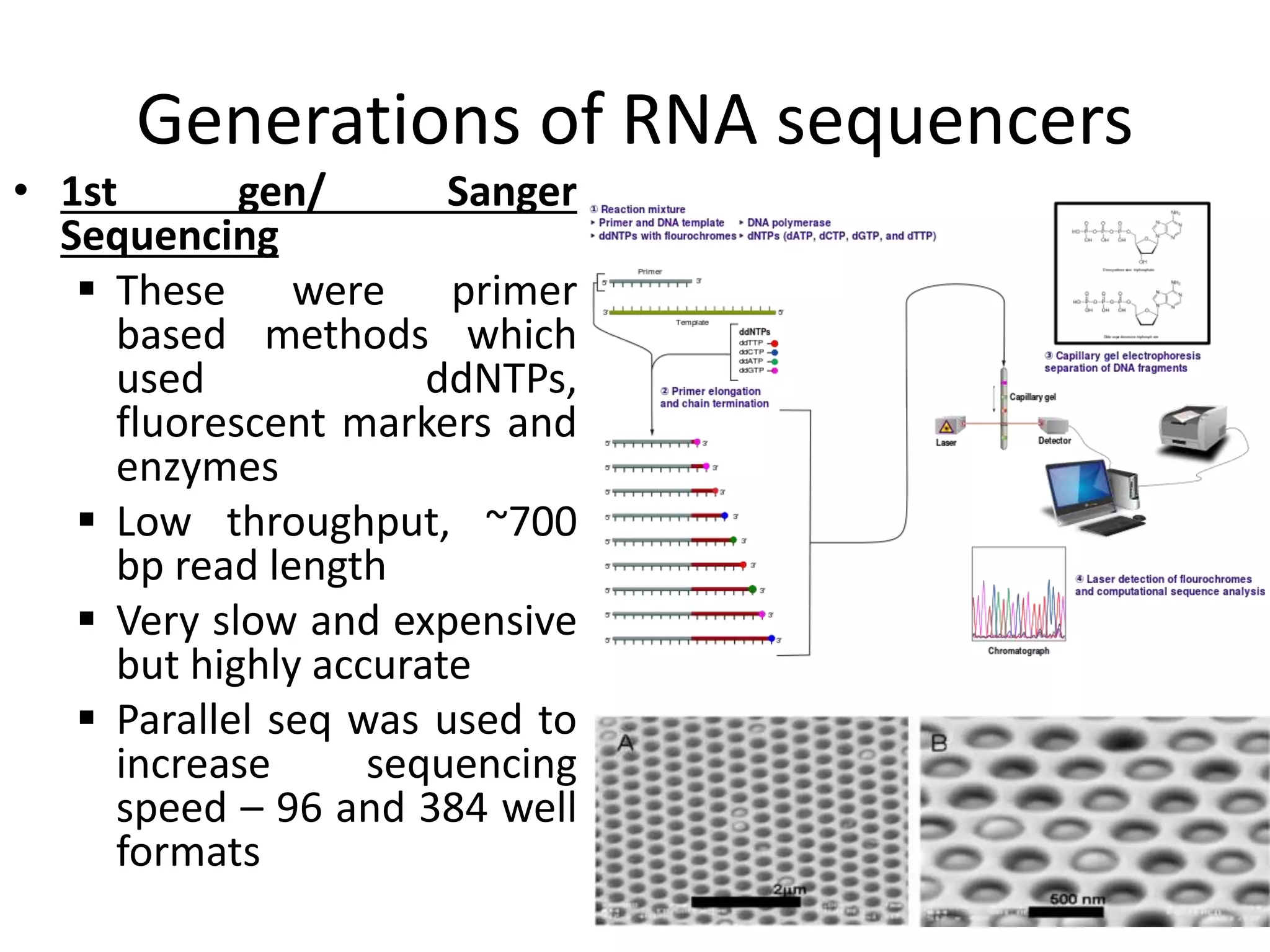

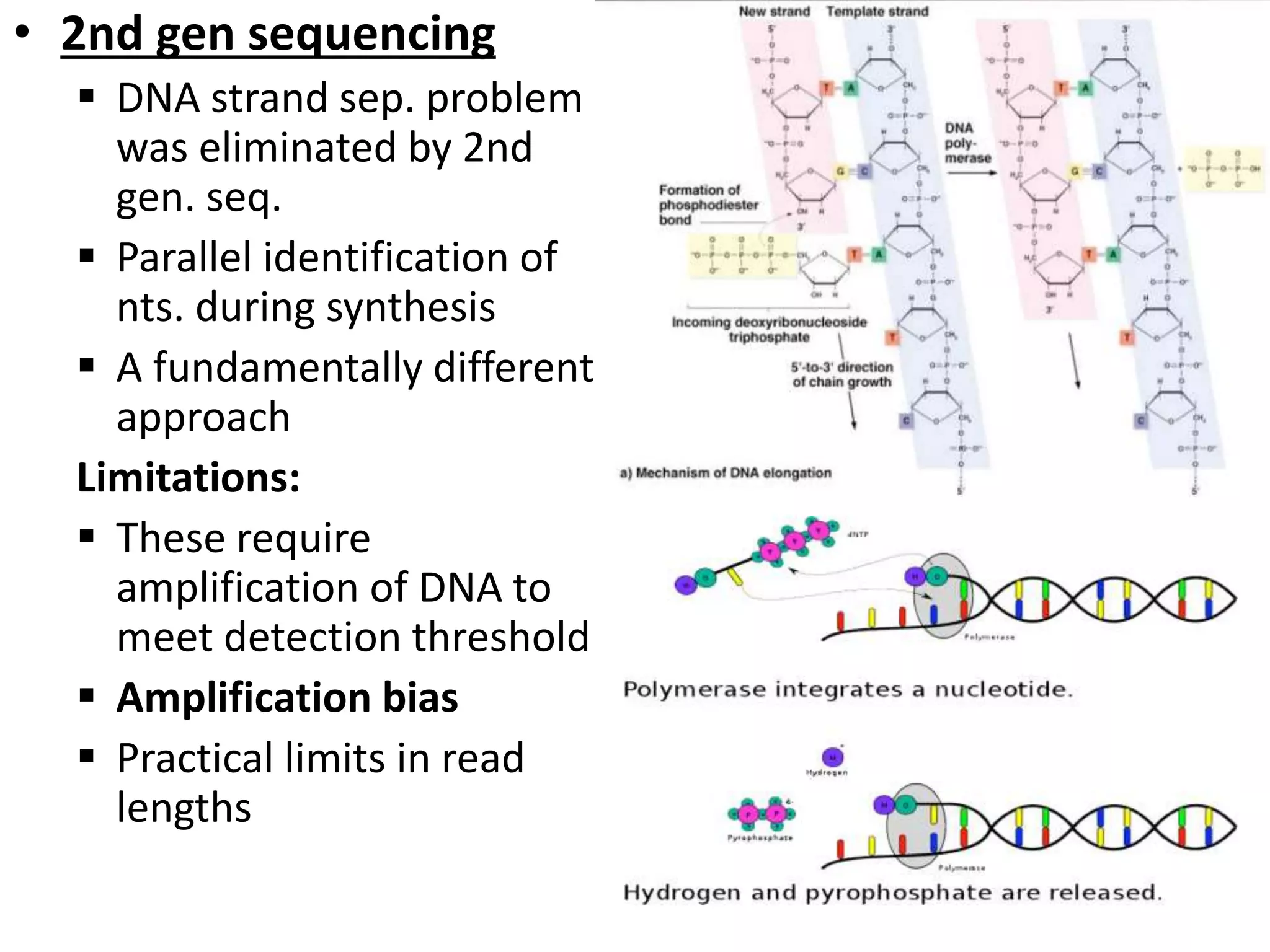
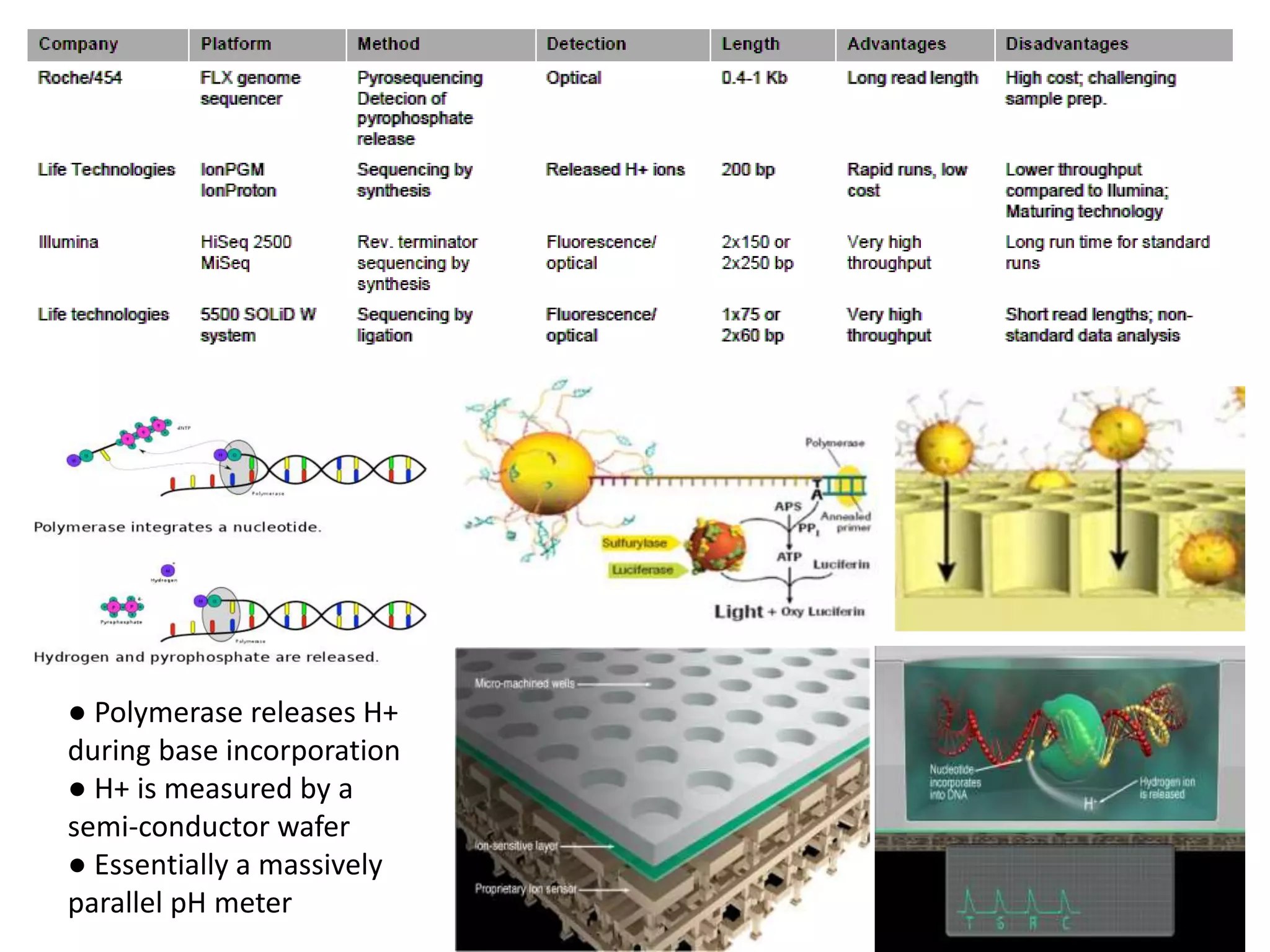
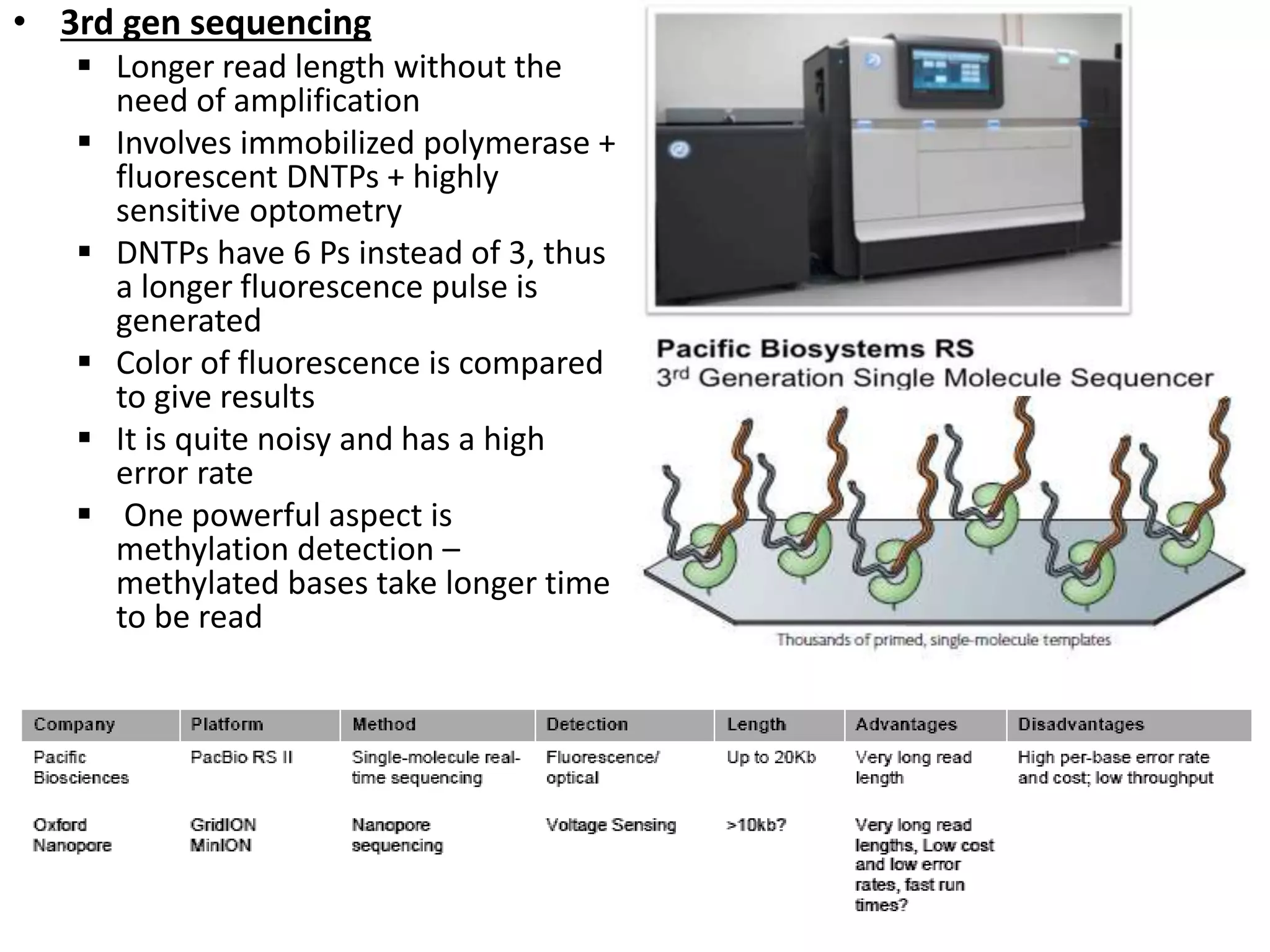
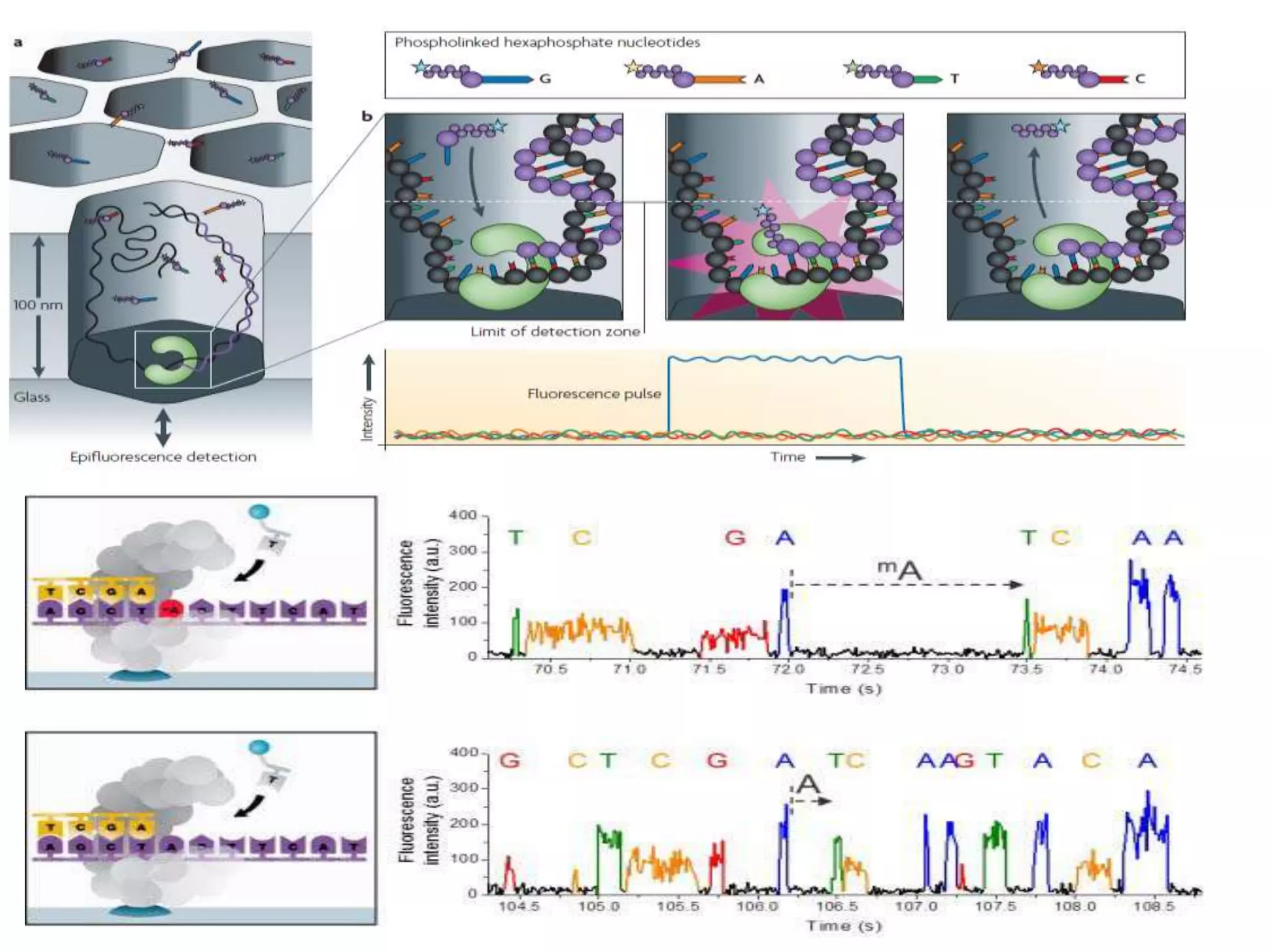

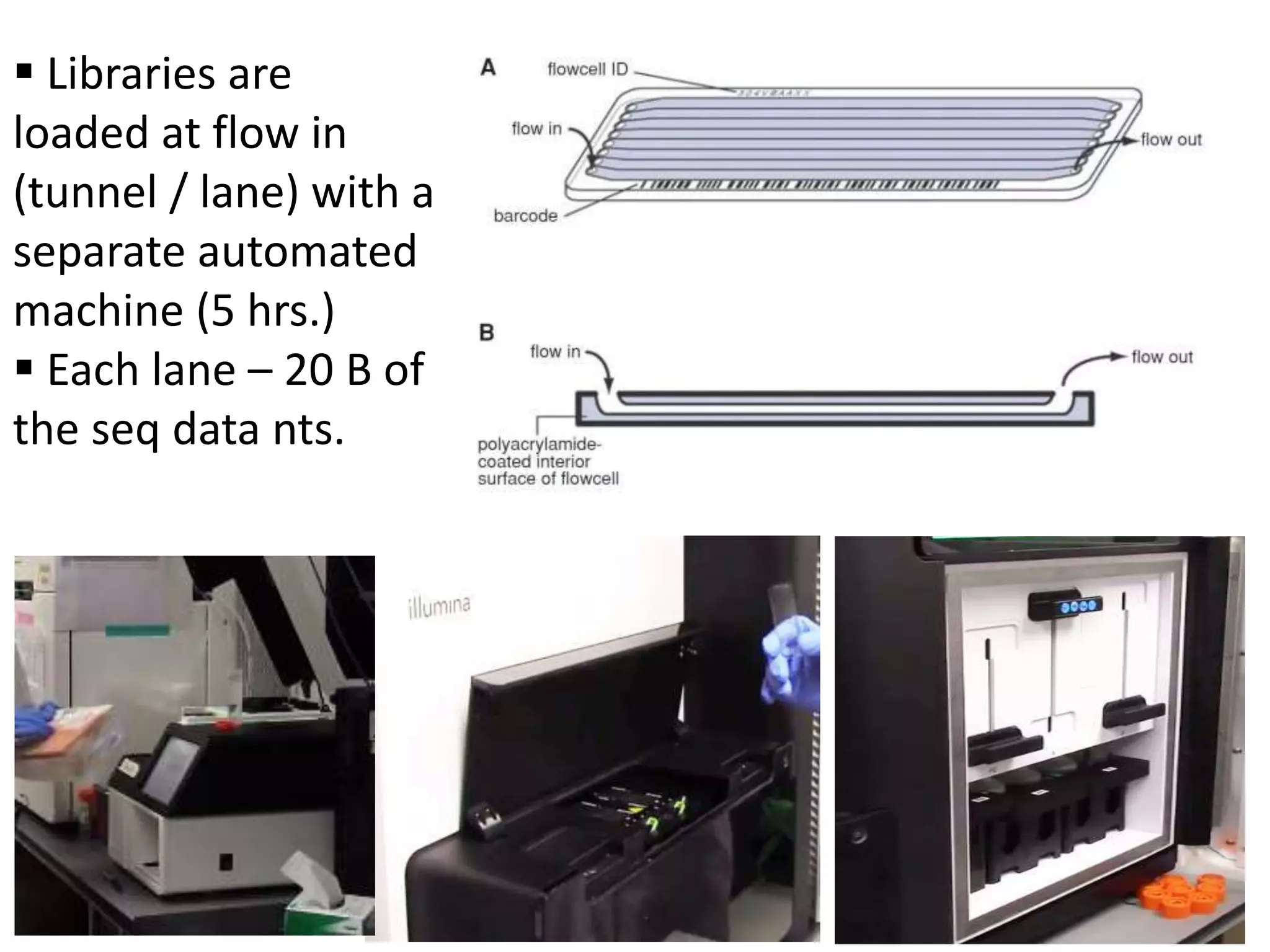
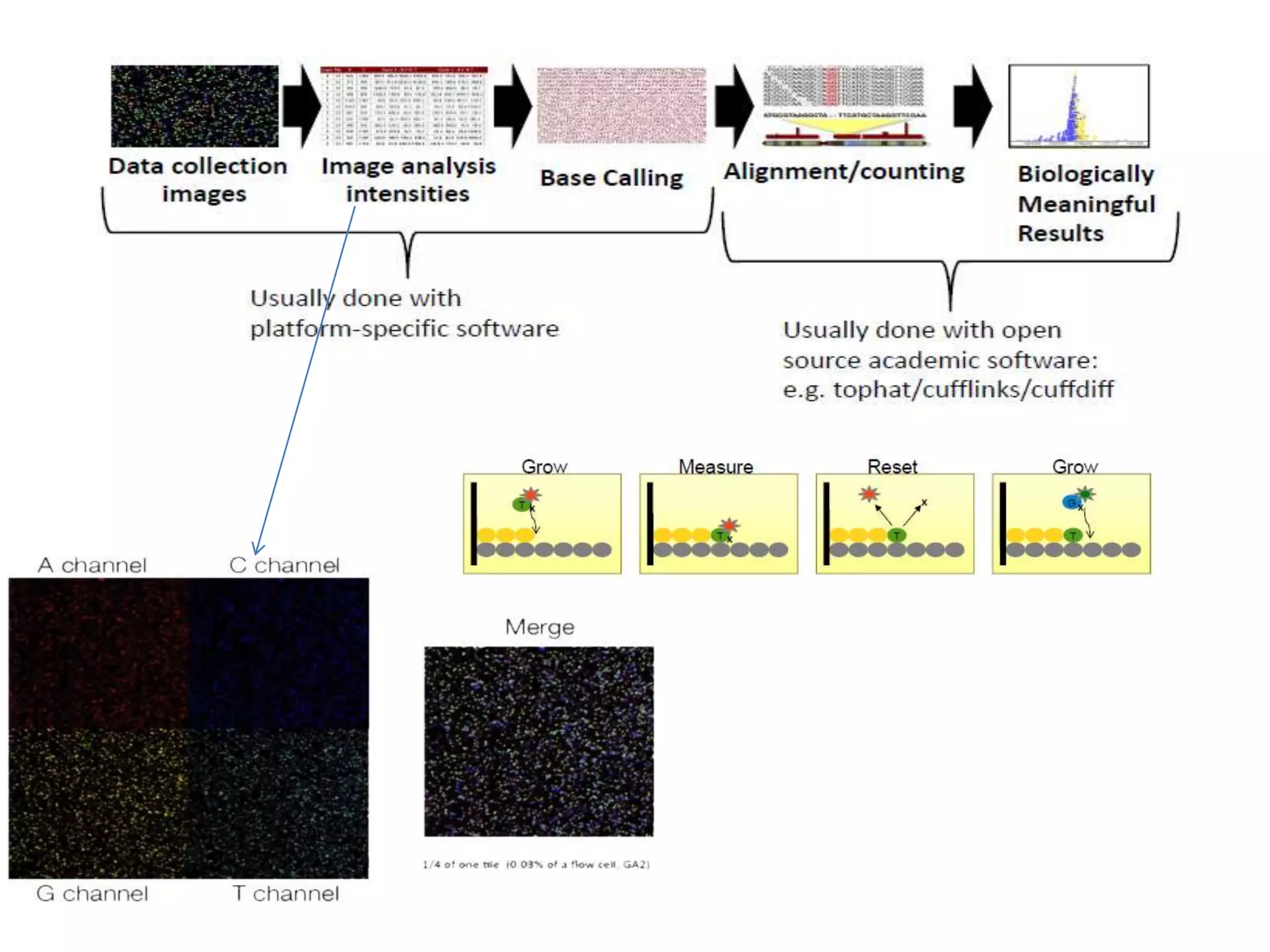
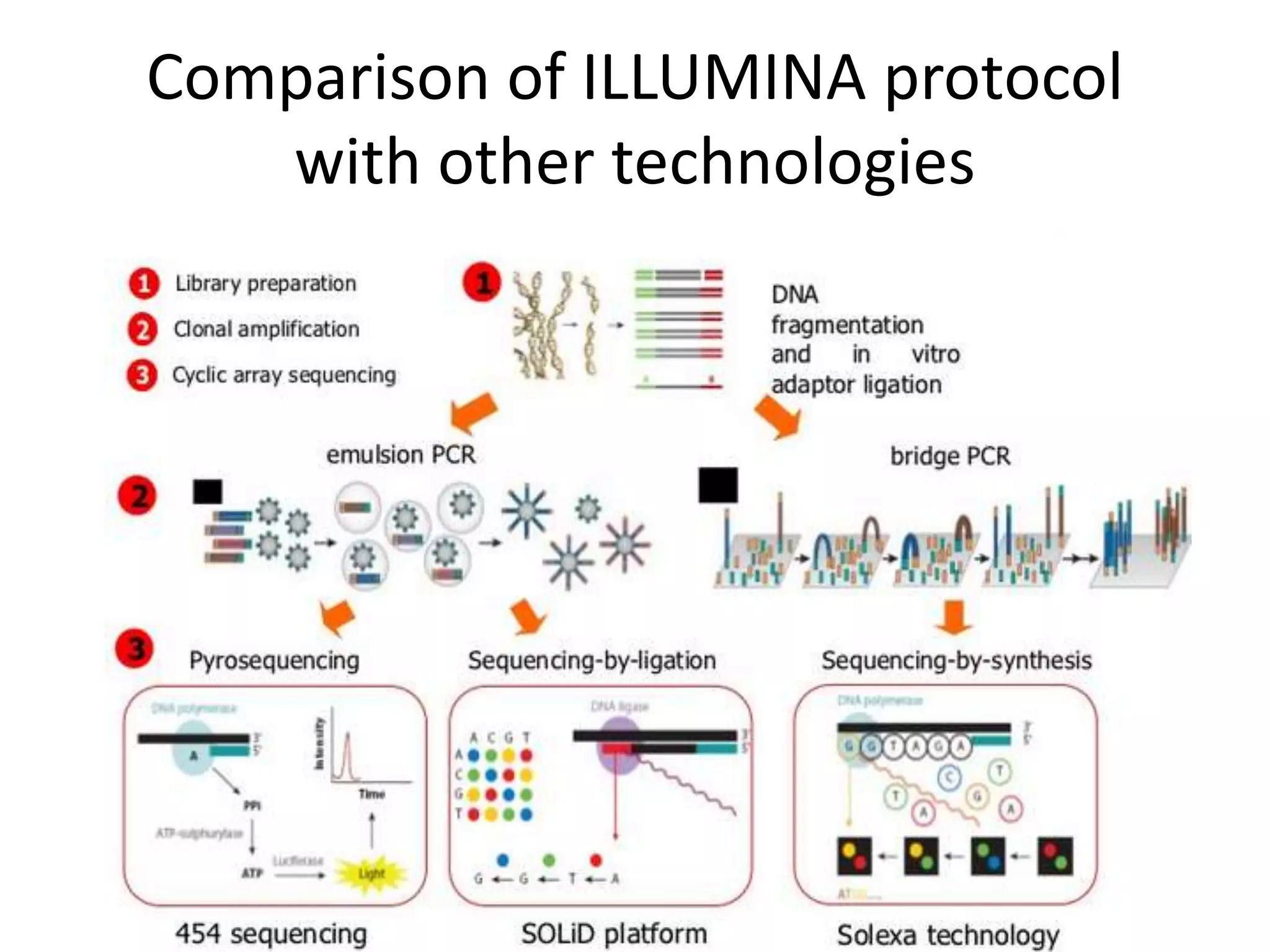
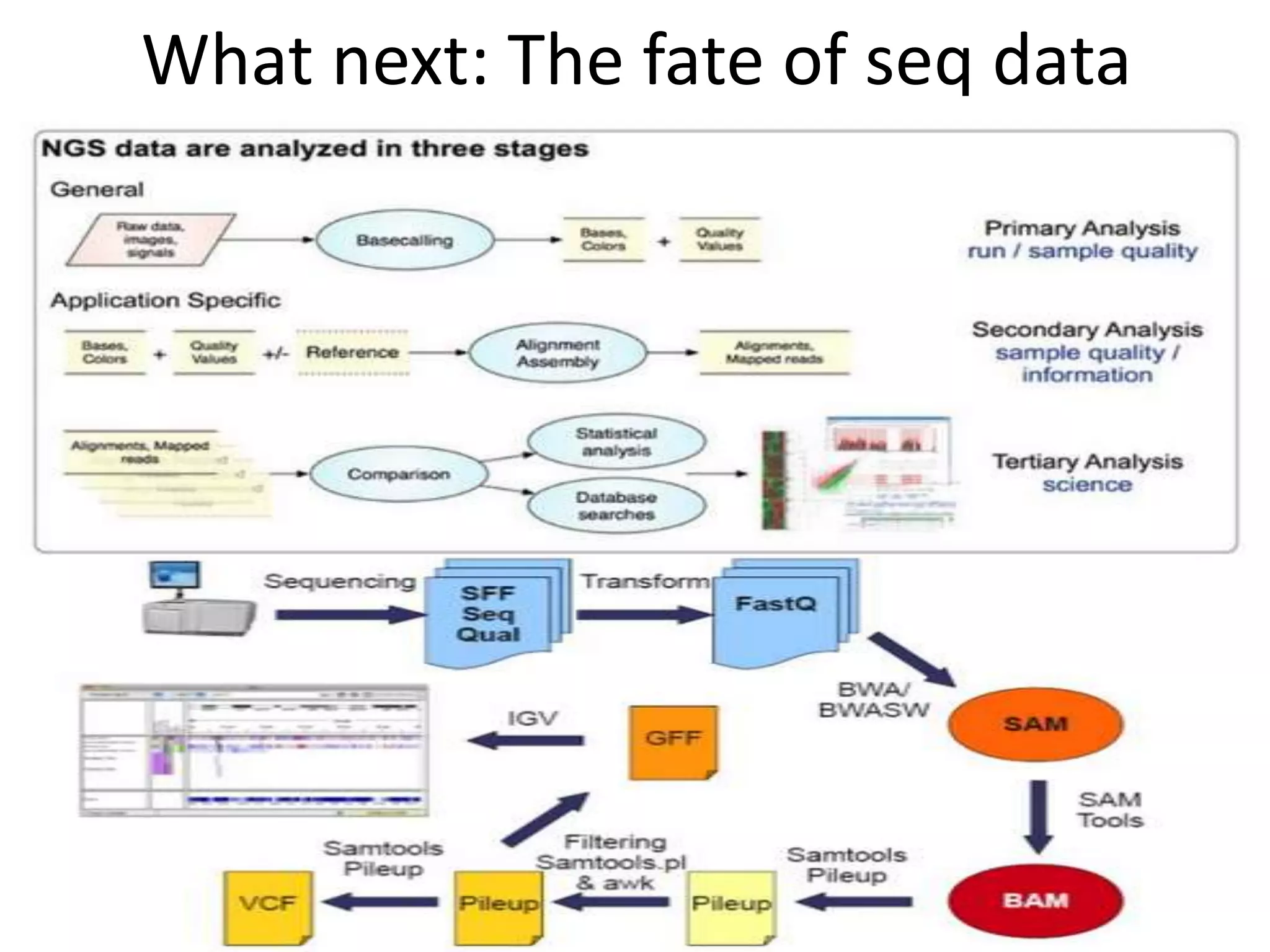

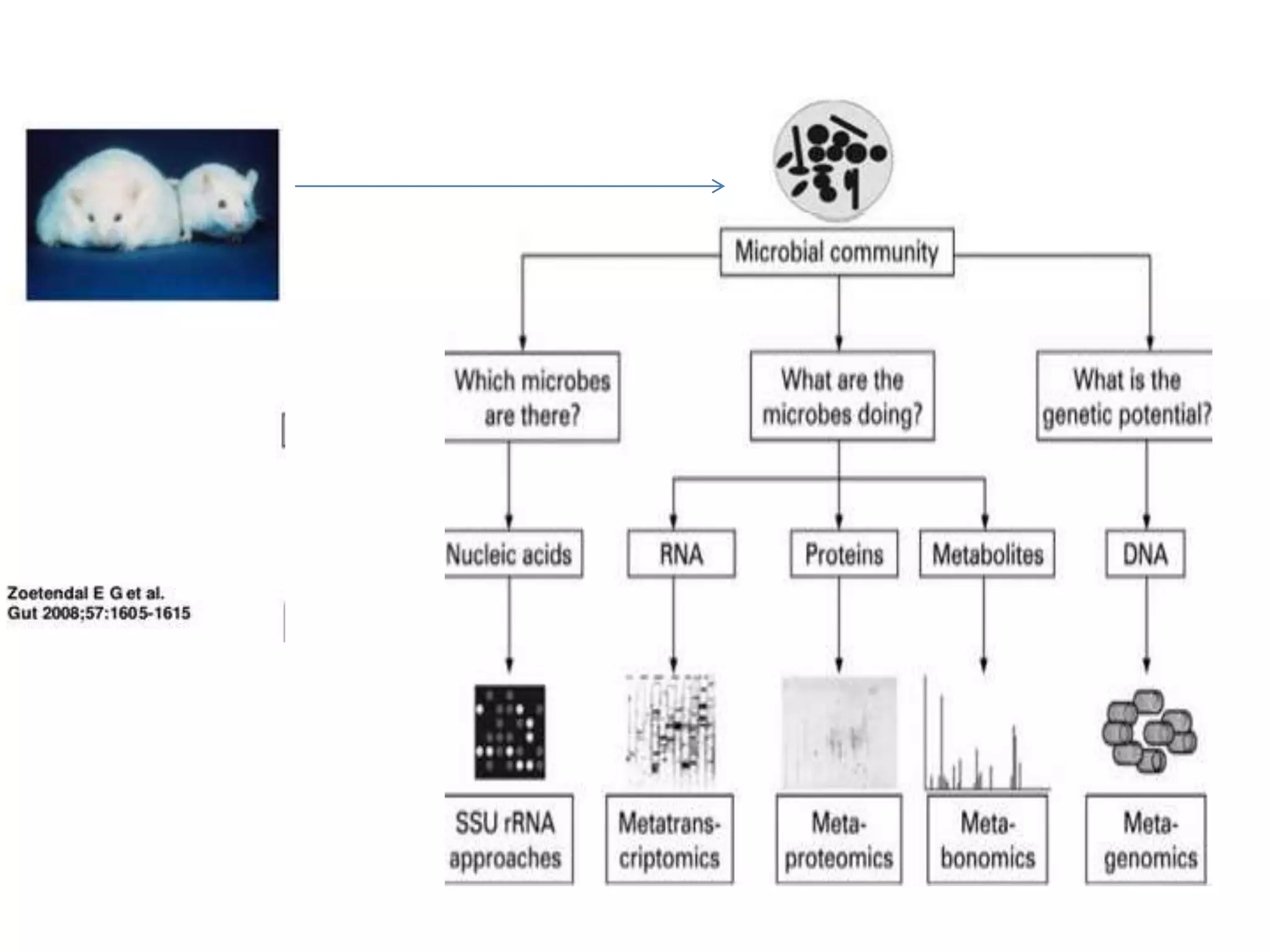





The document discusses next-generation sequencing (NGS) methods, detailing various sequencing technologies and their applications in determining the order of nucleotides in DNA and RNA. It covers topics such as quantitative PCR, library complexity, and the advantages of NGS over traditional Sanger sequencing. The document also highlights recent advancements like the MinION technology and its potential impact on the sequencing market.



































































