Downloaded 37 times

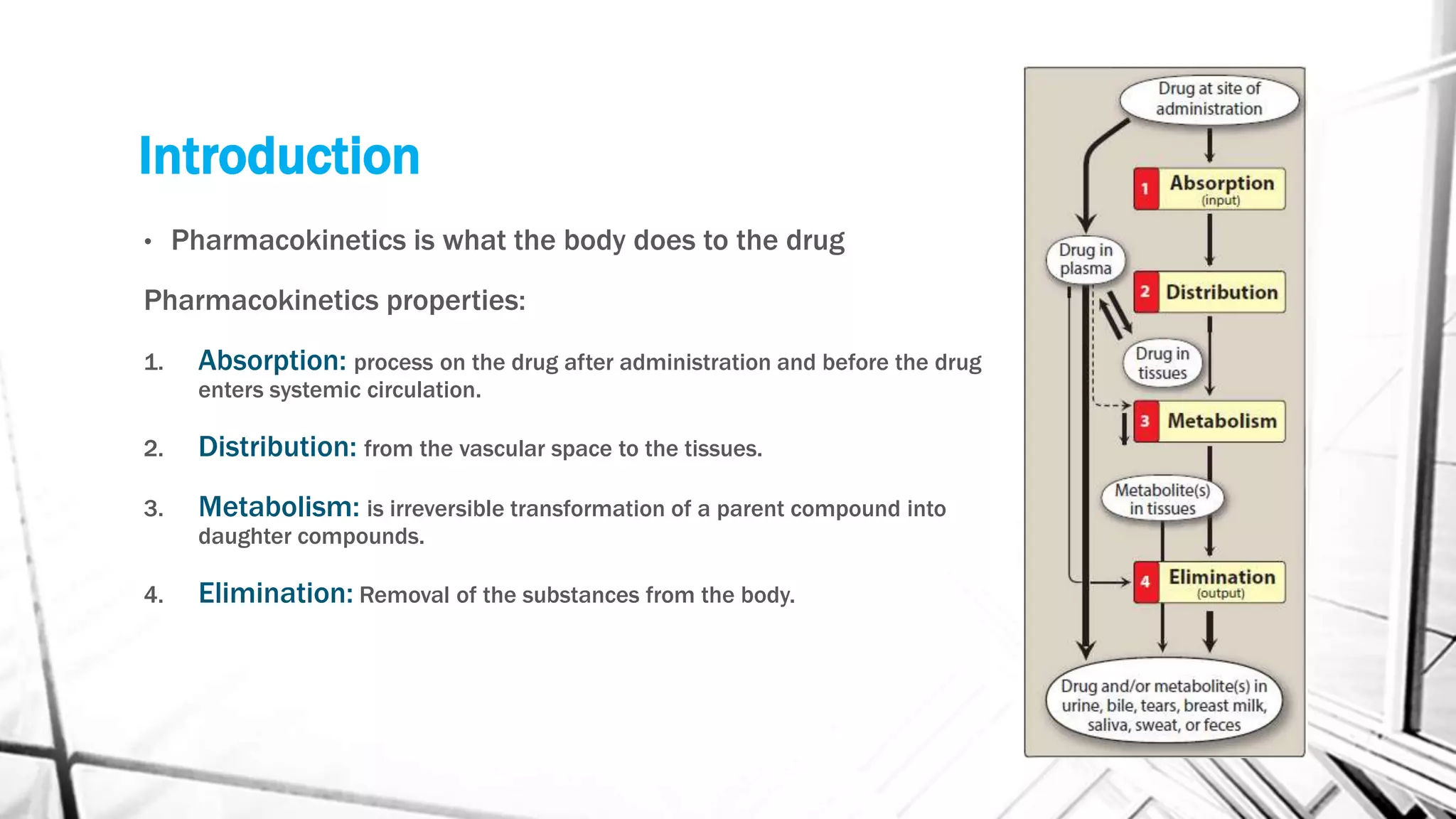






This document discusses the key concepts of pharmacokinetics including absorption, distribution, metabolism, and elimination of drugs in the body. It describes how drugs are absorbed through membranes and distributed to tissues, metabolized through phases I and II reactions, and eliminated from the body through the kidneys, liver, lungs and bile. First and zero order kinetics of drug elimination are also explained, with examples of how the rates of elimination differ between the two models.
















![Clinical Pharmacokinetics-I [half life, order of kinetics, steady state]](https://cdn.slidesharecdn.com/ss_thumbnails/clinicalpk-ihalflifeorderofkineticssteadystate-140217020044-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)






































































