This document provides an overview of informed consent, including its purpose, basic principles, elements, documentation requirements, and application to special populations. Key points include:
- Informed consent is a process of communication between researchers and participants to ensure participants understand research risks/benefits before agreeing to participate.
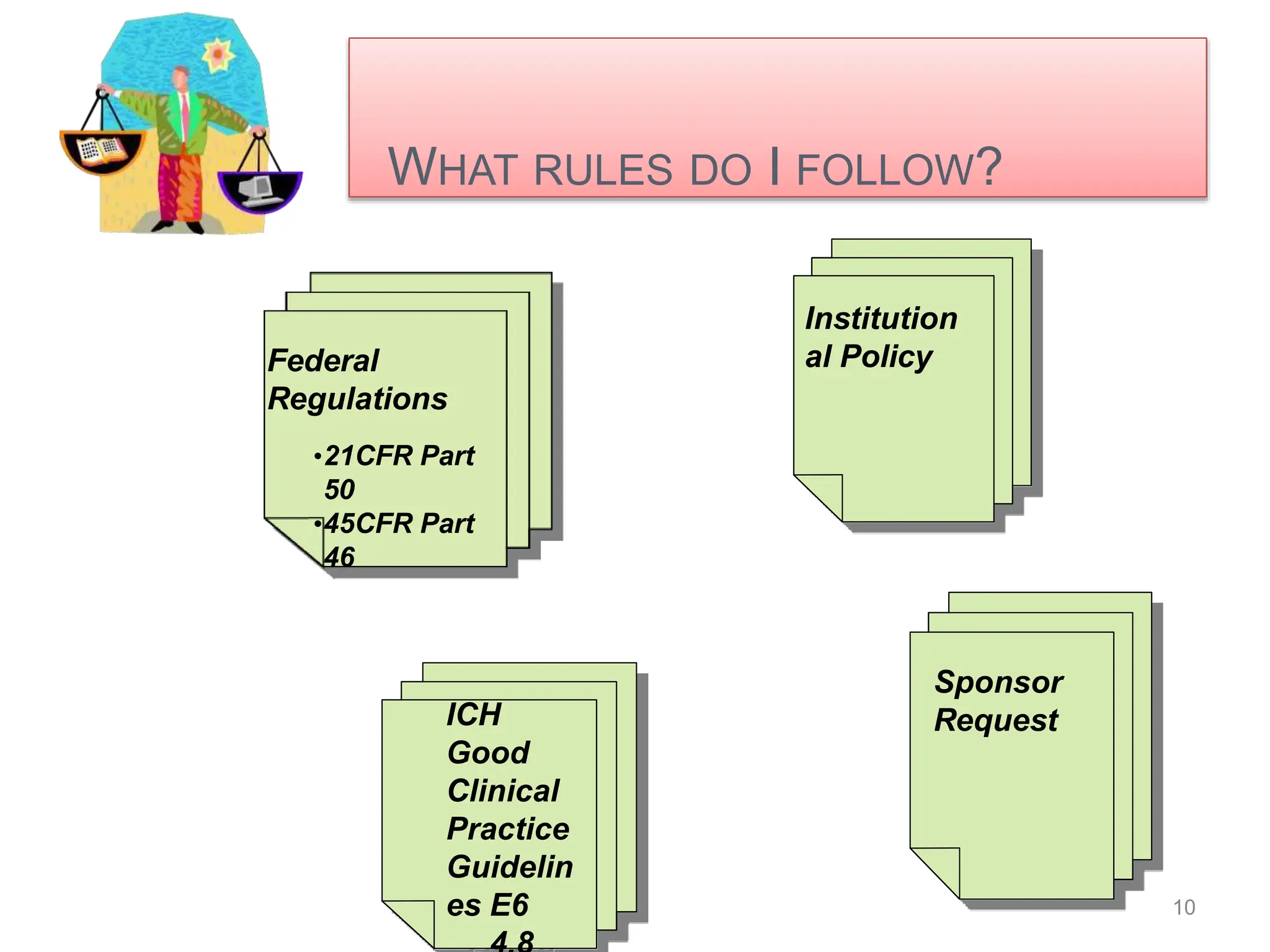
- The basic principles of informed consent are autonomy, beneficence, and justice. Informed consent forms must include study details, risks/benefits, confidentiality, and participant responsibilities.
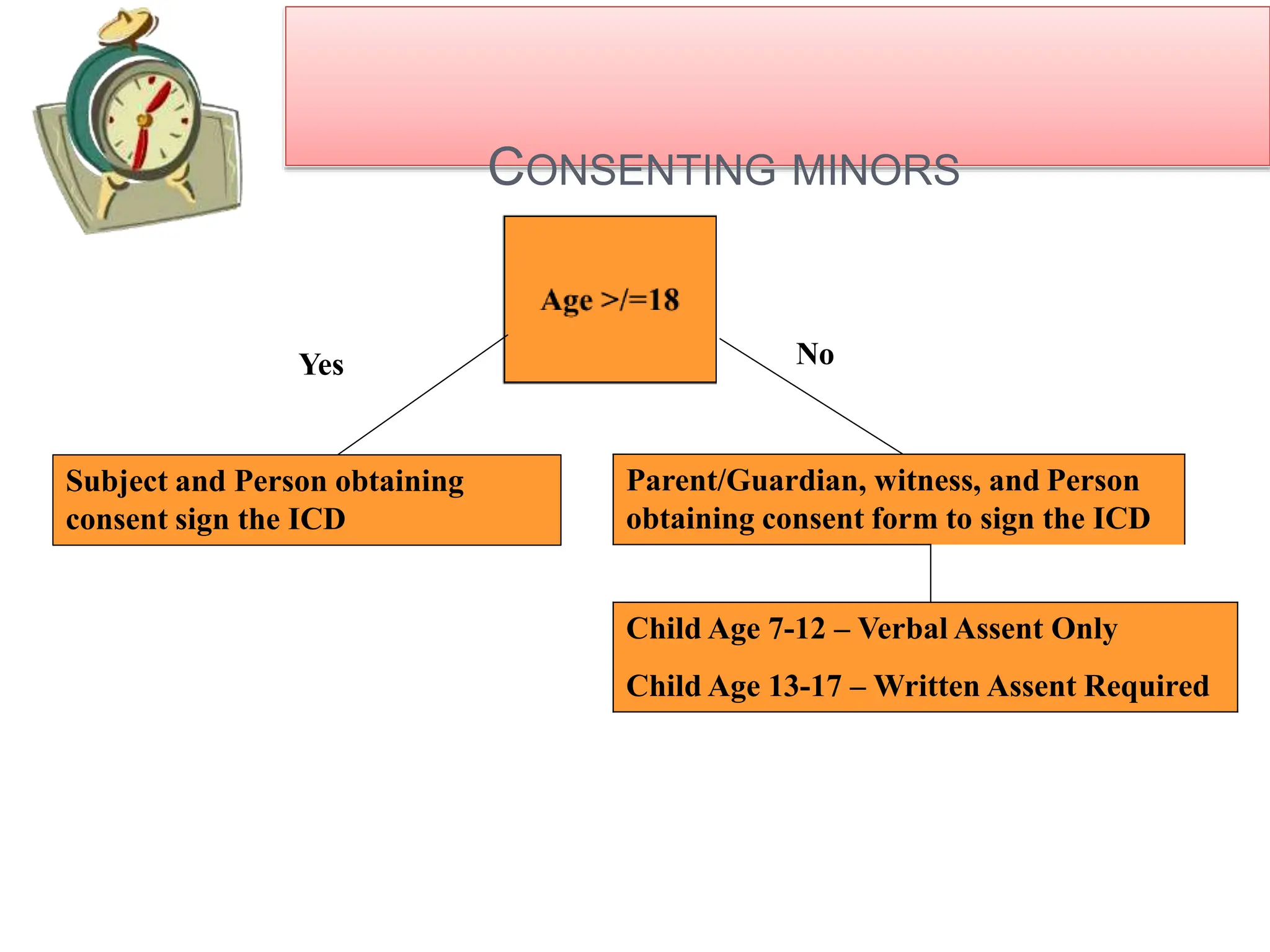
- Special considerations are given for obtaining consent from children, pregnant women, non-English speakers, and individuals with impaired decision-making capacity. Revisions to the consent form require IRB approval and re-consenting of participants