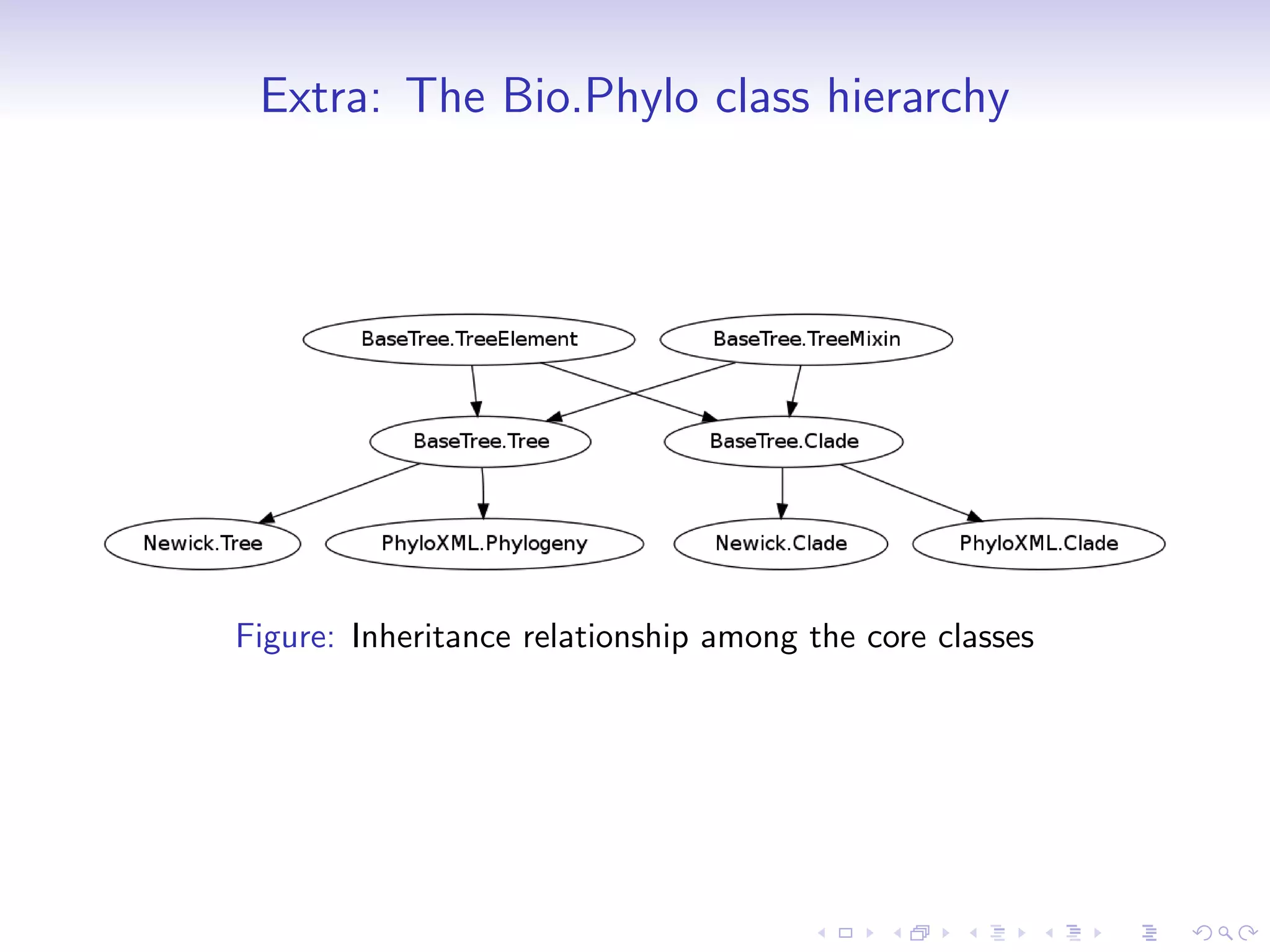
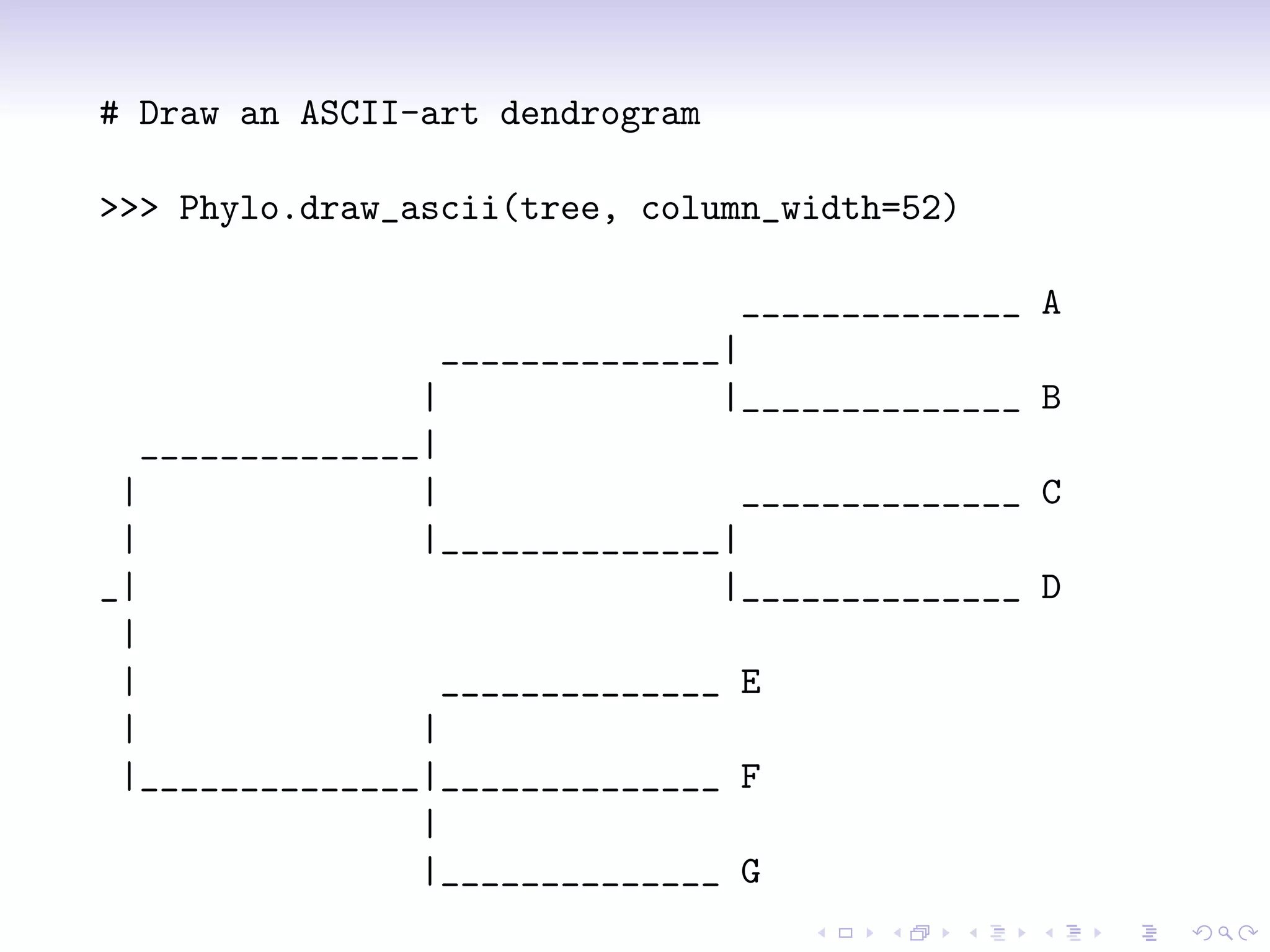
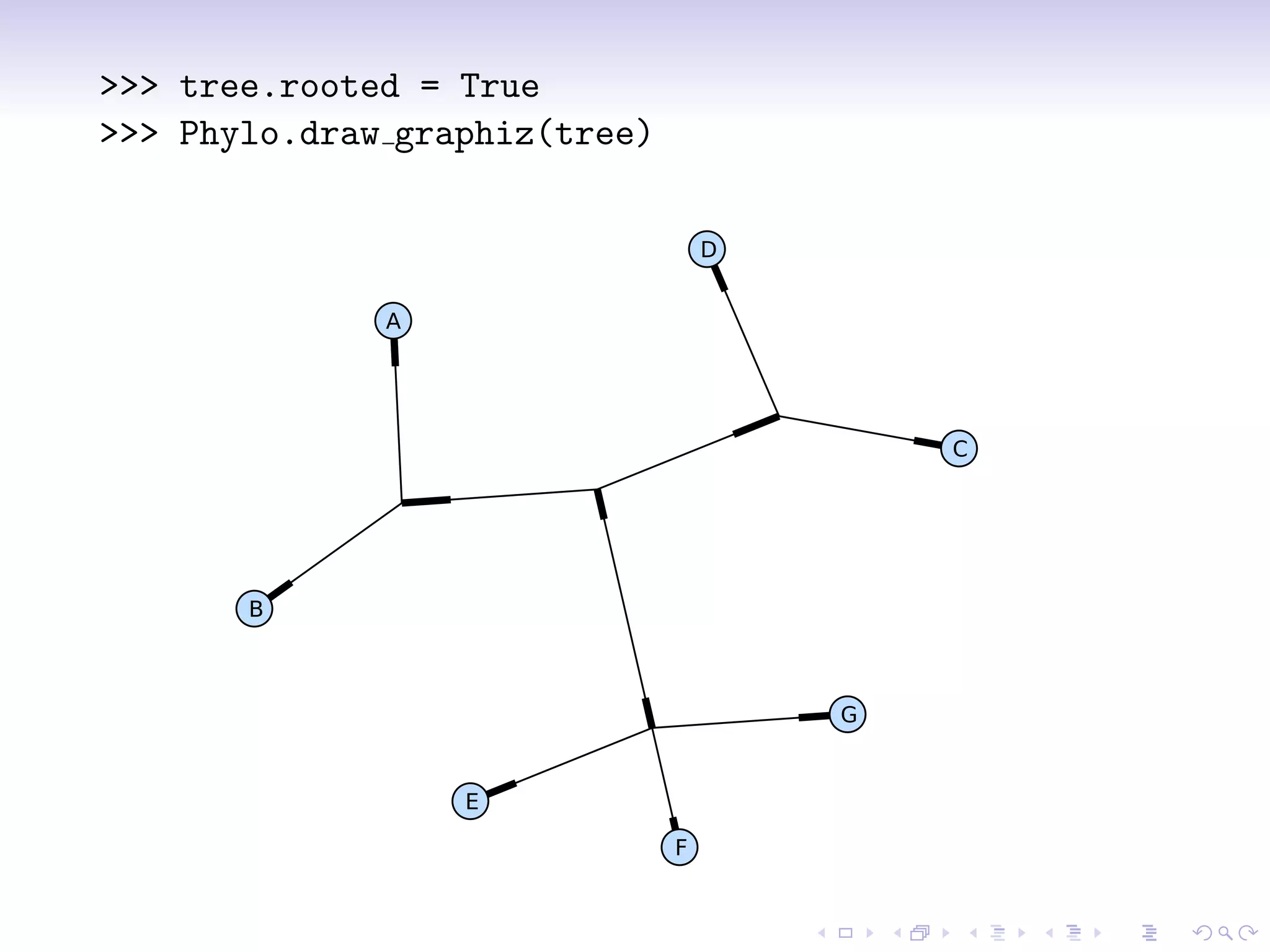
Bio.phylo is a new phylogenetics toolkit for Biopython that enables exploration, modification, and visualization of phylogenetic trees while supporting various file formats like Newick and phyloXML. It includes features for reading, annotating, and visually representing trees, as well as promoting trees to more informative formats, such as converting a Newick tree to phyloXML. The document outlines example usage, methods, and available classes for enhancing phylogenetic analysis.







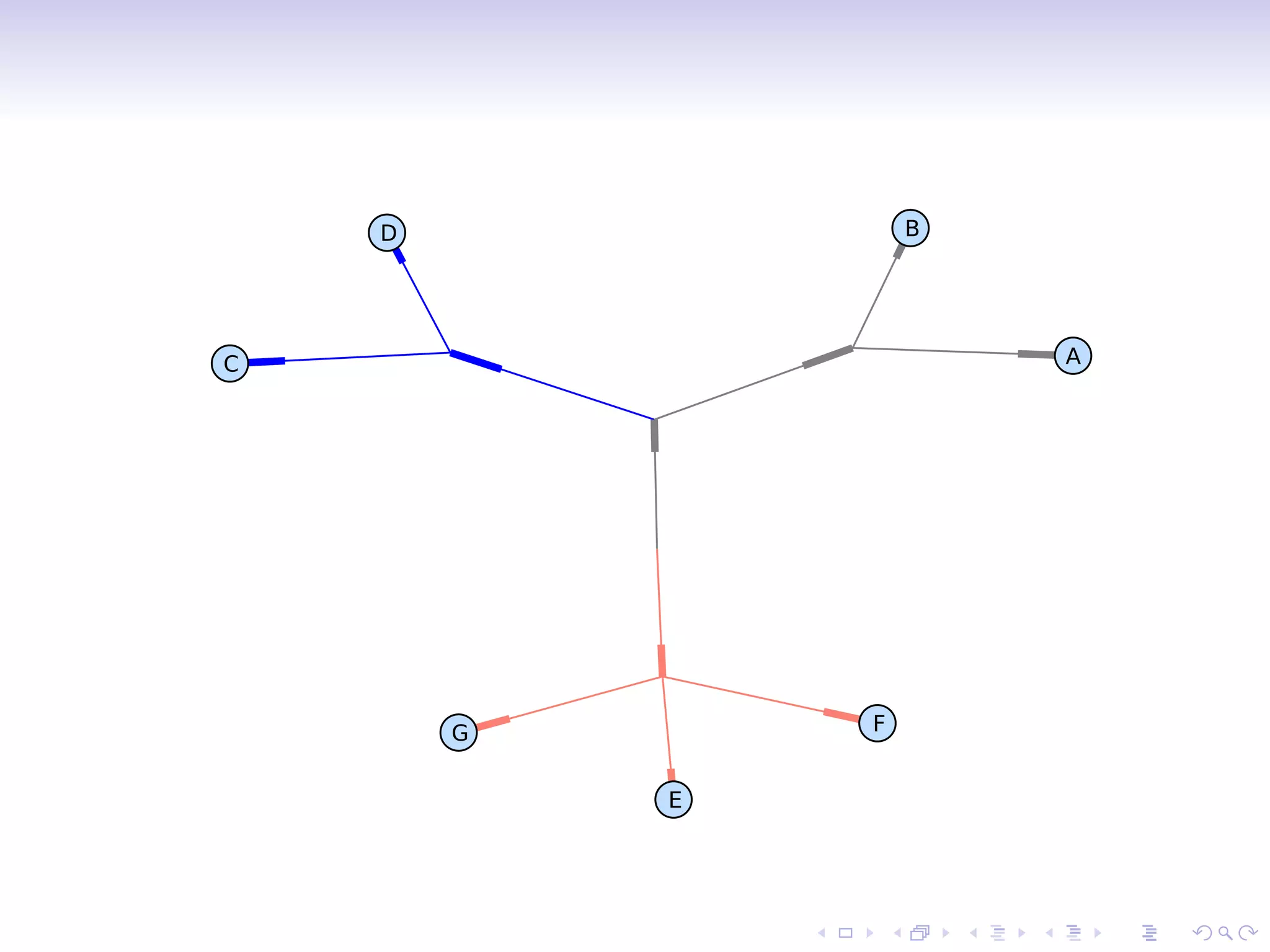
![Branch color
>>> phy.root.color = (128, 128, 128)
Or:
>>> phy.root.color = ’#808080’
Or:
>>> phy.root.color = ’gray’
Find clades by attribute values:
>>> mrca = phy.common ancestor({’name’:’E’},
{’name’:’F’})
>>> mrca.color = ’salmon’
Directly index a clade:
>>> phy.clade[0,1].color = ’blue’
>>> Phylo.draw graphviz(phy, prog=’neato’)](https://image.slidesharecdn.com/bosc2010-biophylo-talevich-100721205437-phpapp01/75/Bio-Phylo-Phylogenetics-in-Biopython-BOSC-2010-10-2048.jpg)