Downloaded 633 times

























The document presents a detailed guide on the investigation and preprocessing of raw RNA-seq data, emphasizing the importance of experimental design, sequencing options, and quality control methods. It discusses the differences between single-end and paired-end sequencing, the relevance of strandedness, and the consequences of technical and biological contaminations in the data. Preprocessing steps such as removing low-quality and contaminant sequences are critical for accurate differential expression analysis and successful mapping to reference genomes.
Presentation on the raw data investigation process in sequencing, by Joachim Jacob.
How to decide on the number of samples and depth for reliable statistics; emphasizes pilot data and sequencing options.
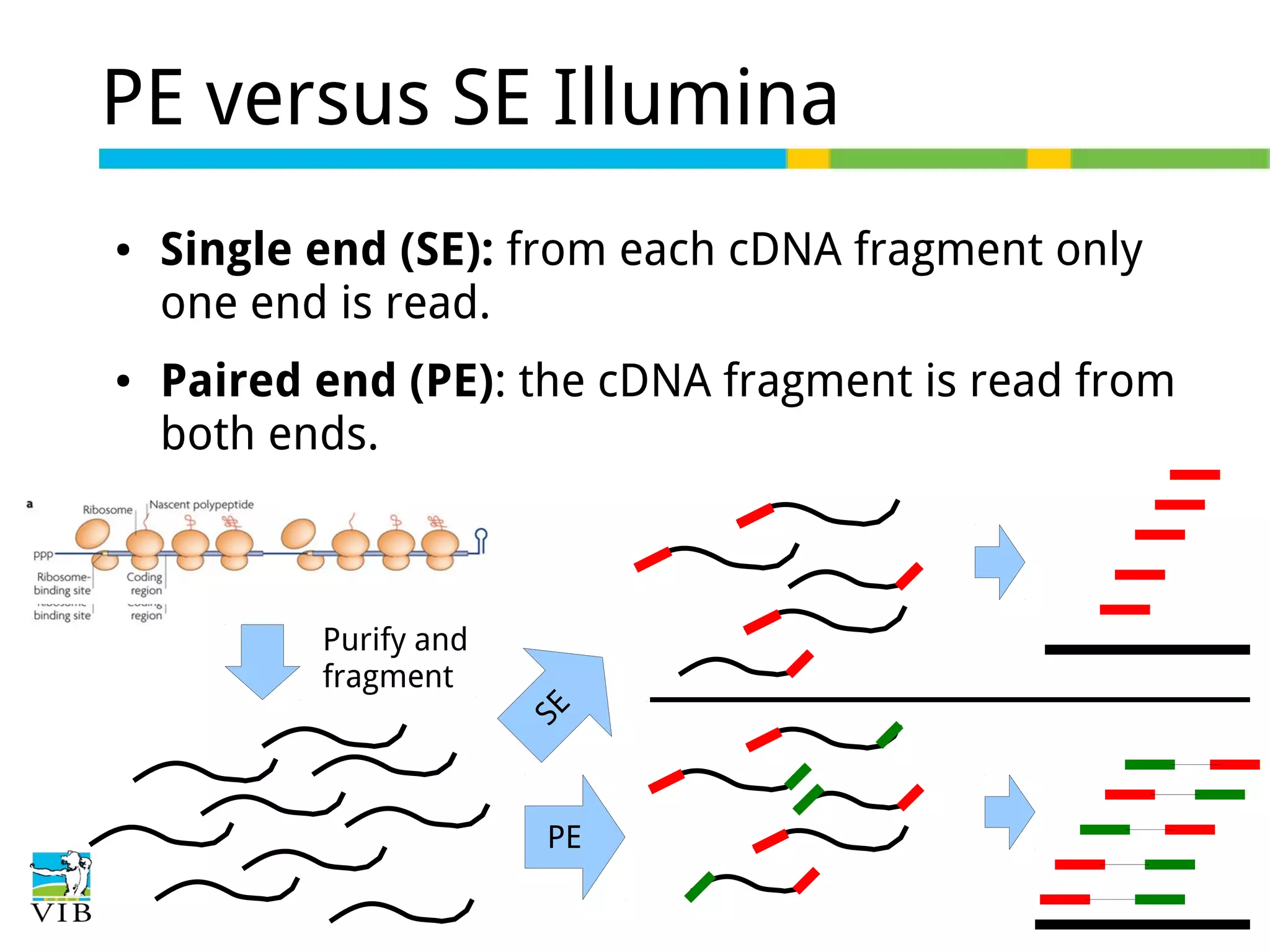
Differences between single-end (SE) and paired-end (PE) sequencing; PE aids in splice junction detection and transcriptome assembly.
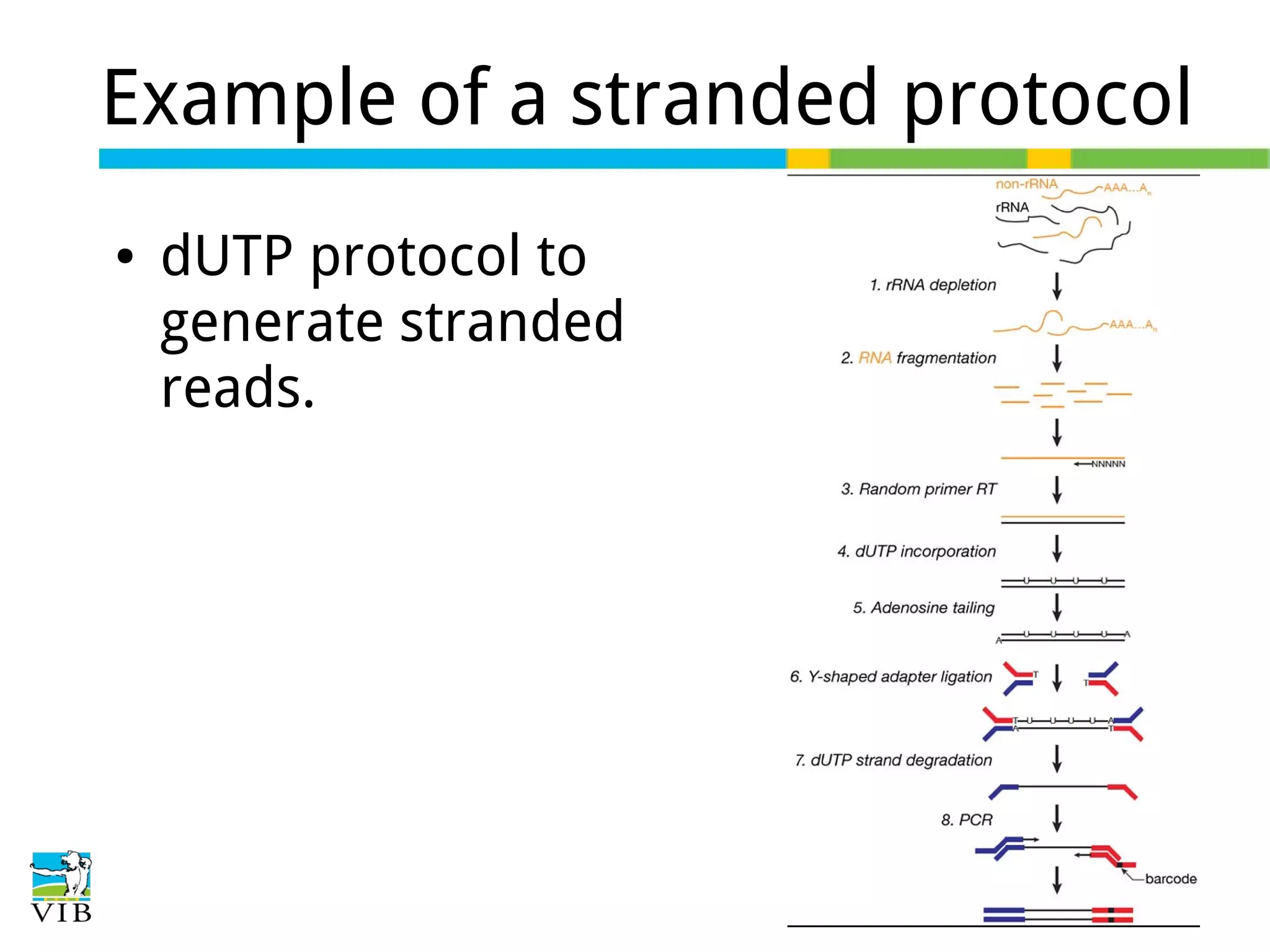
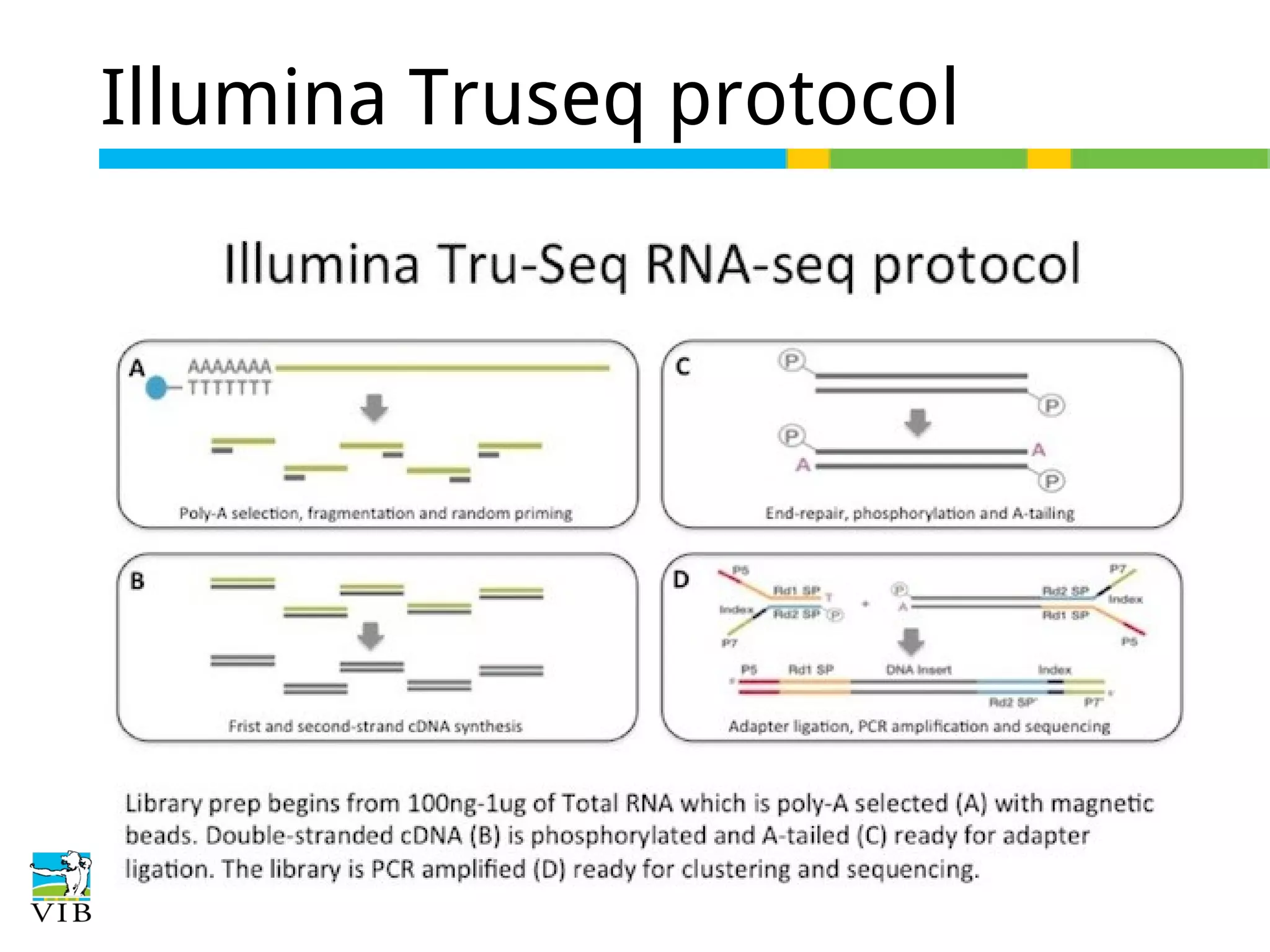
Stranded protocols generate reads corresponding to sense or antisense strands, influencing read counts and accuracy, especially for overlapping genes.
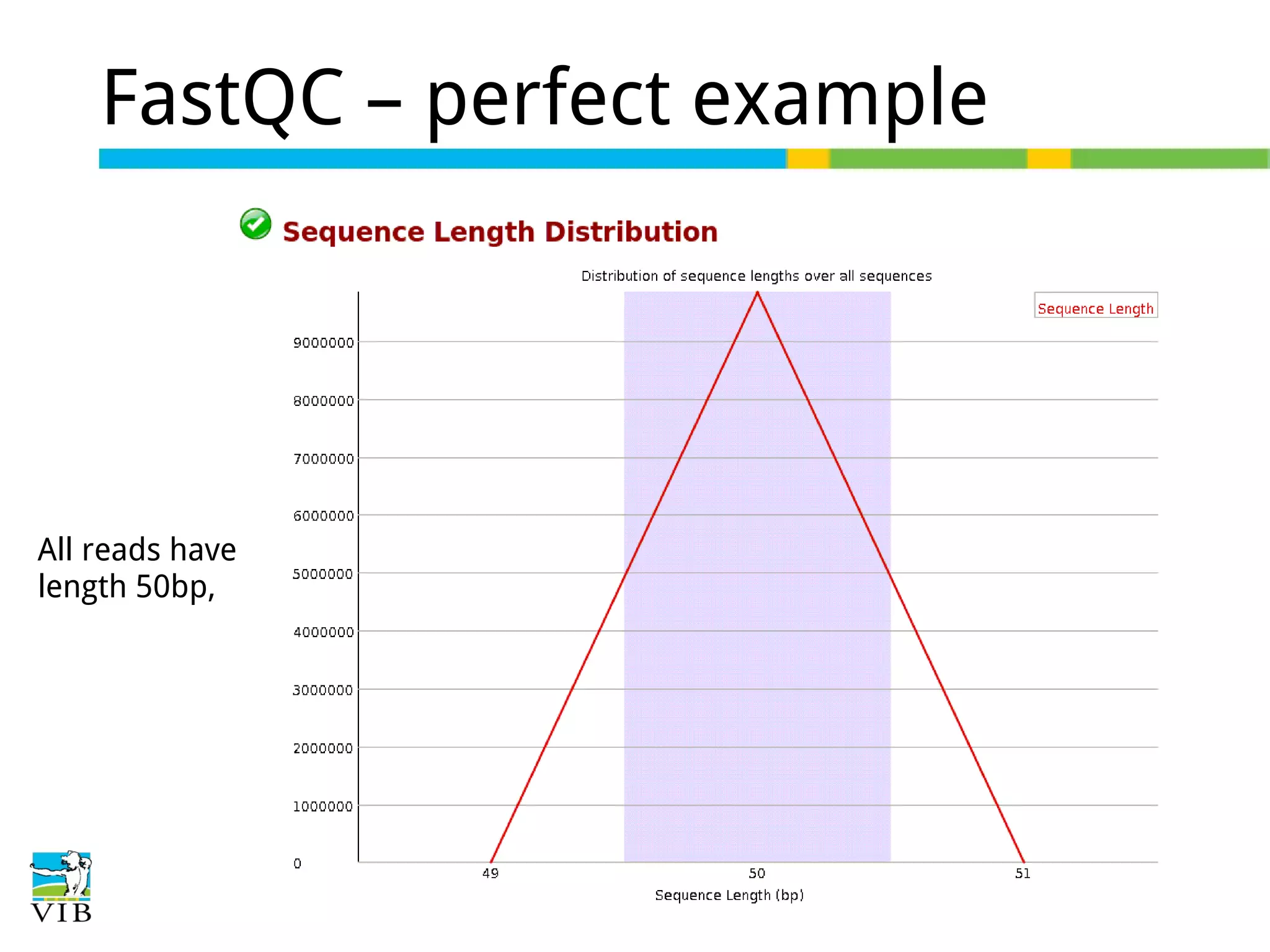
Read length (e.g., 50 bp) affects alignment precision; it is less crucial for quantification but important for transcriptome assembly.
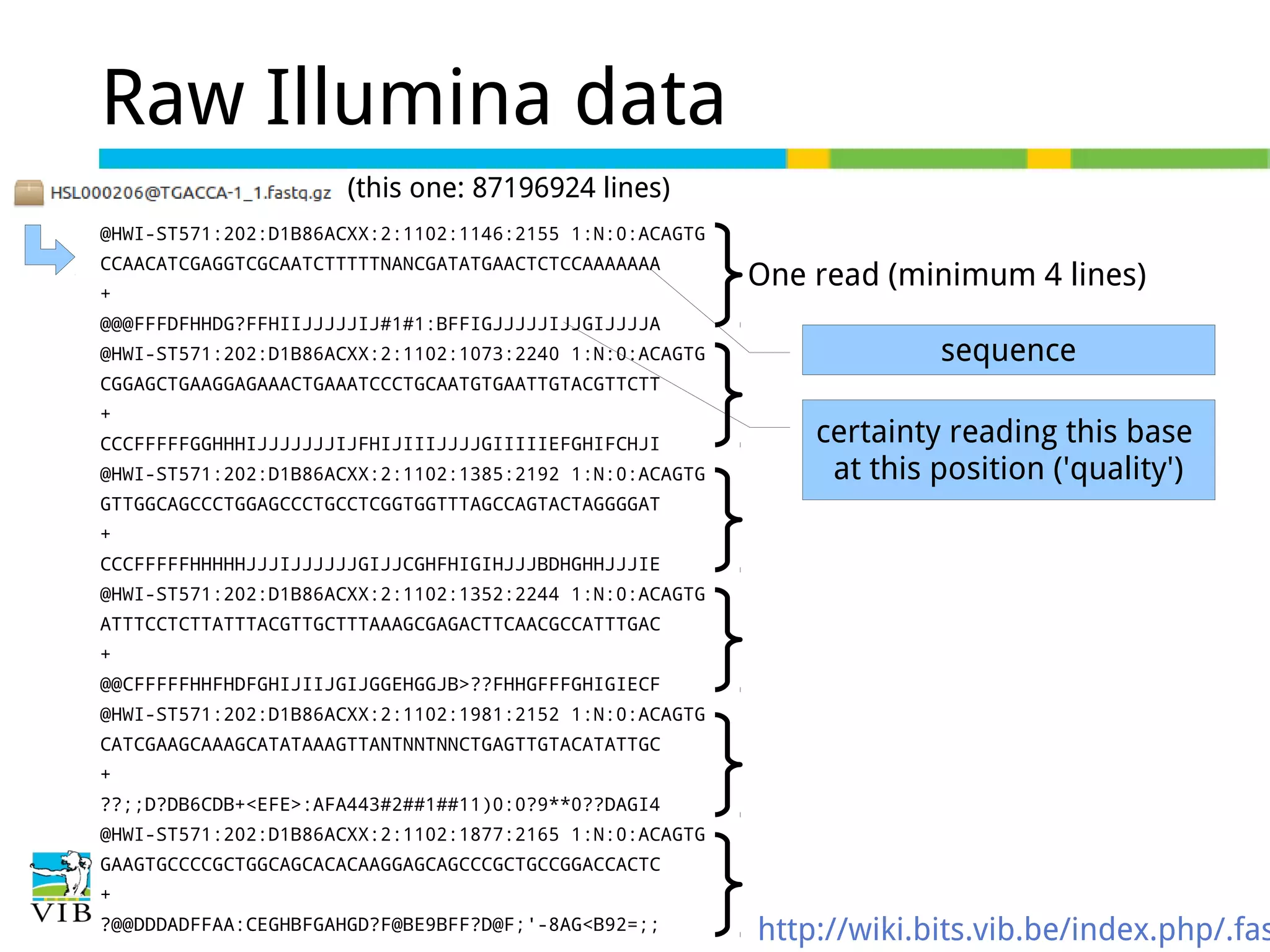
Illustrates the format of raw Illumina data, including barcodes, quality scores, and sequence representation.
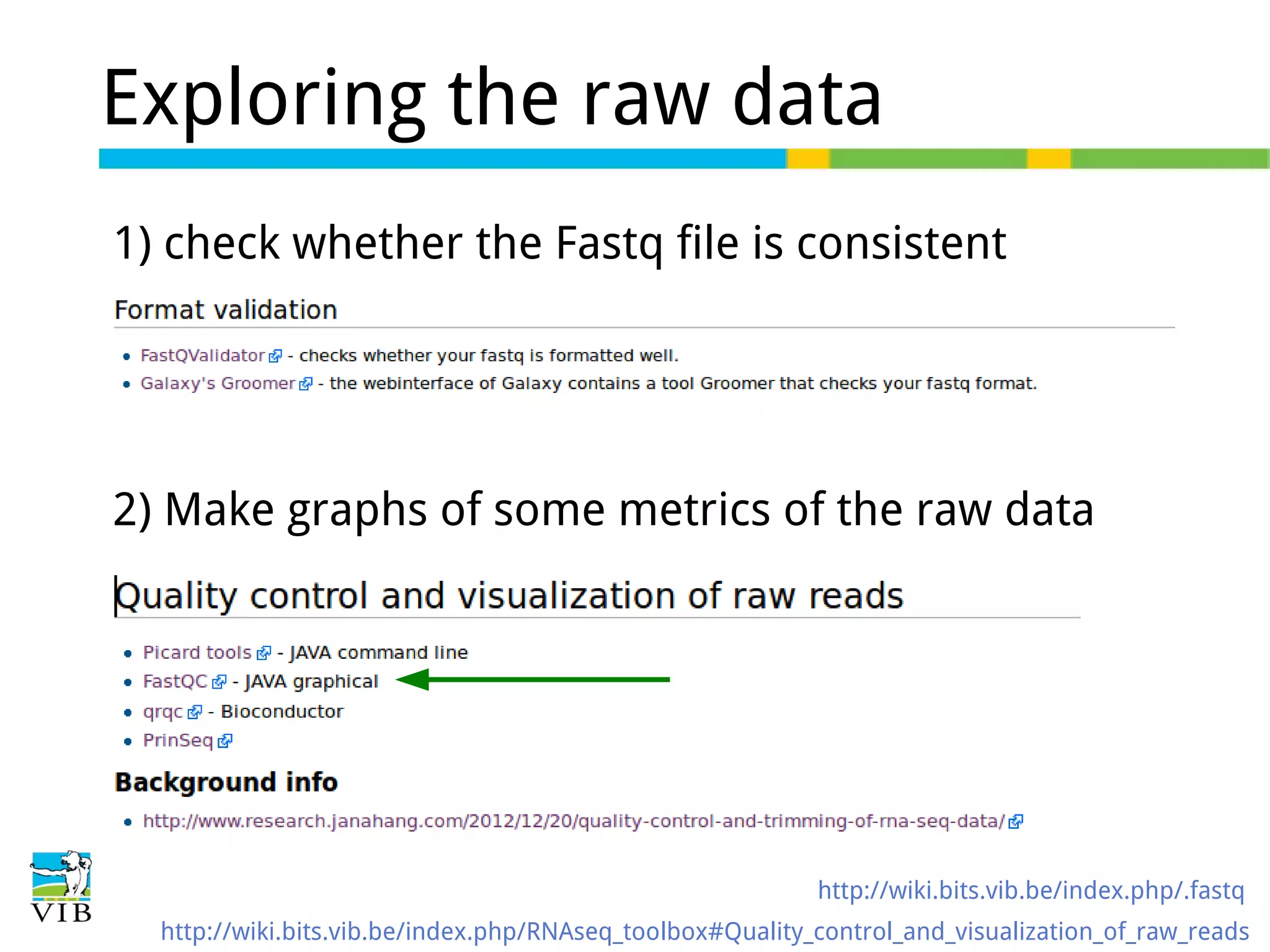
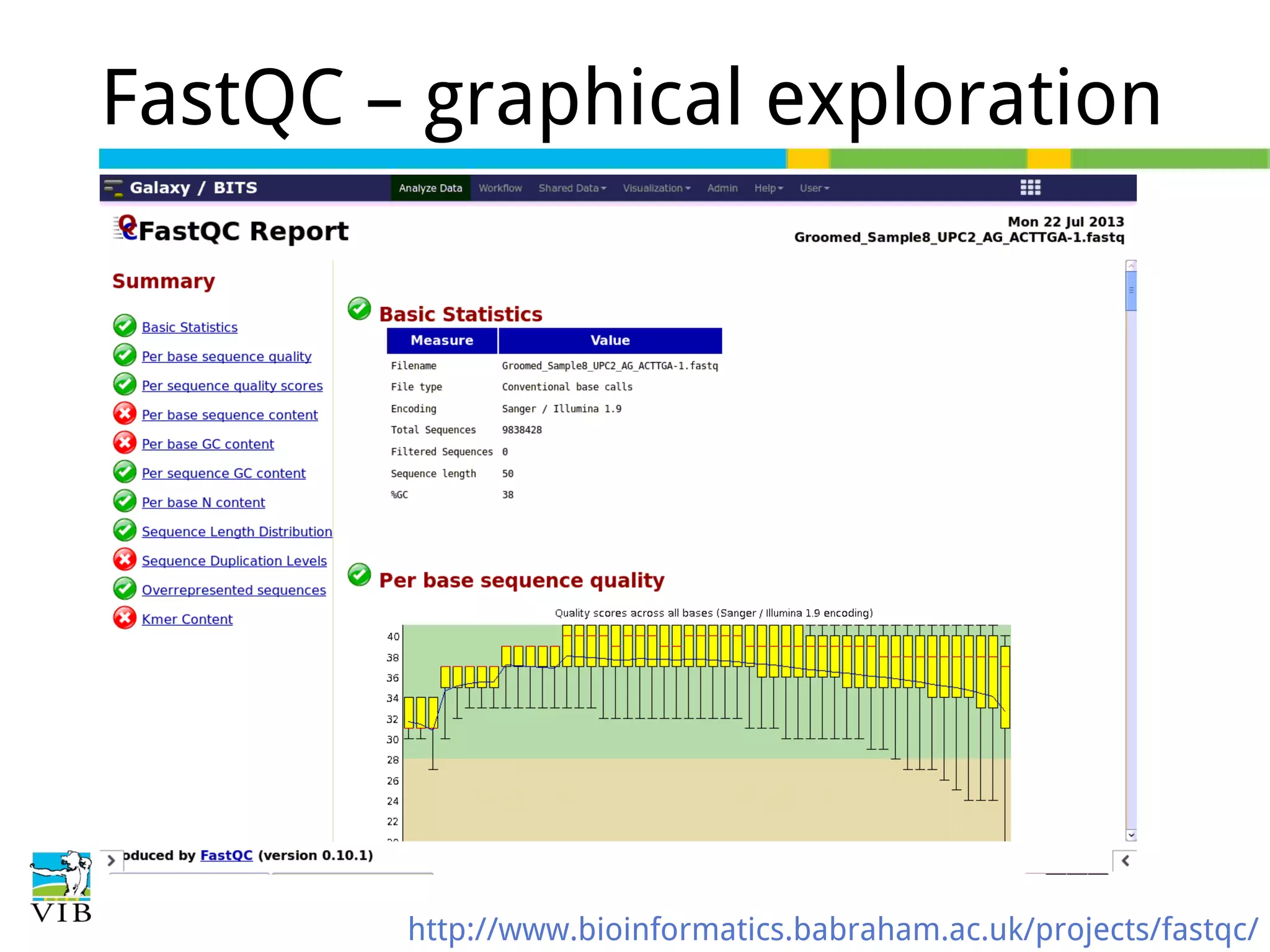
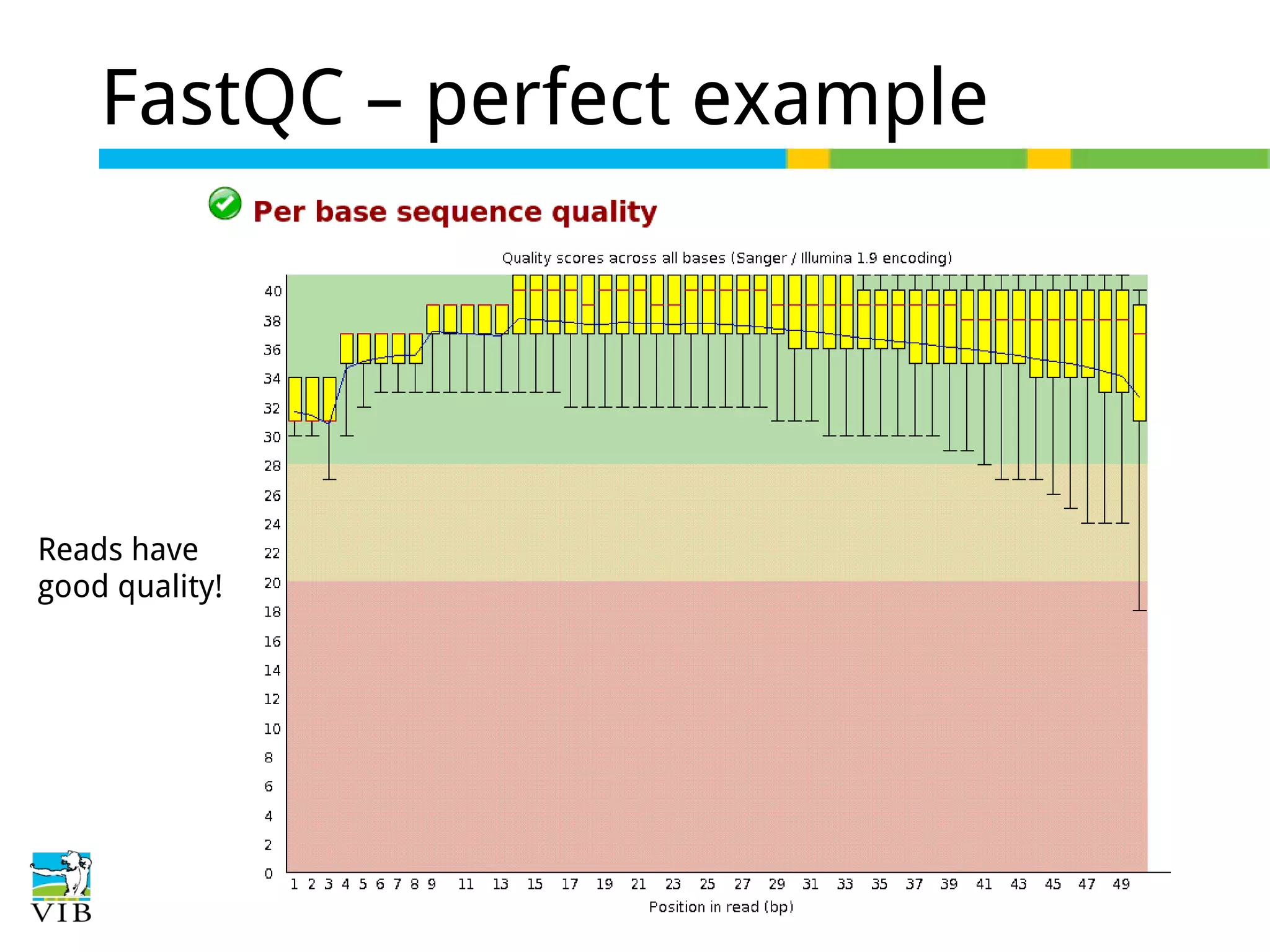
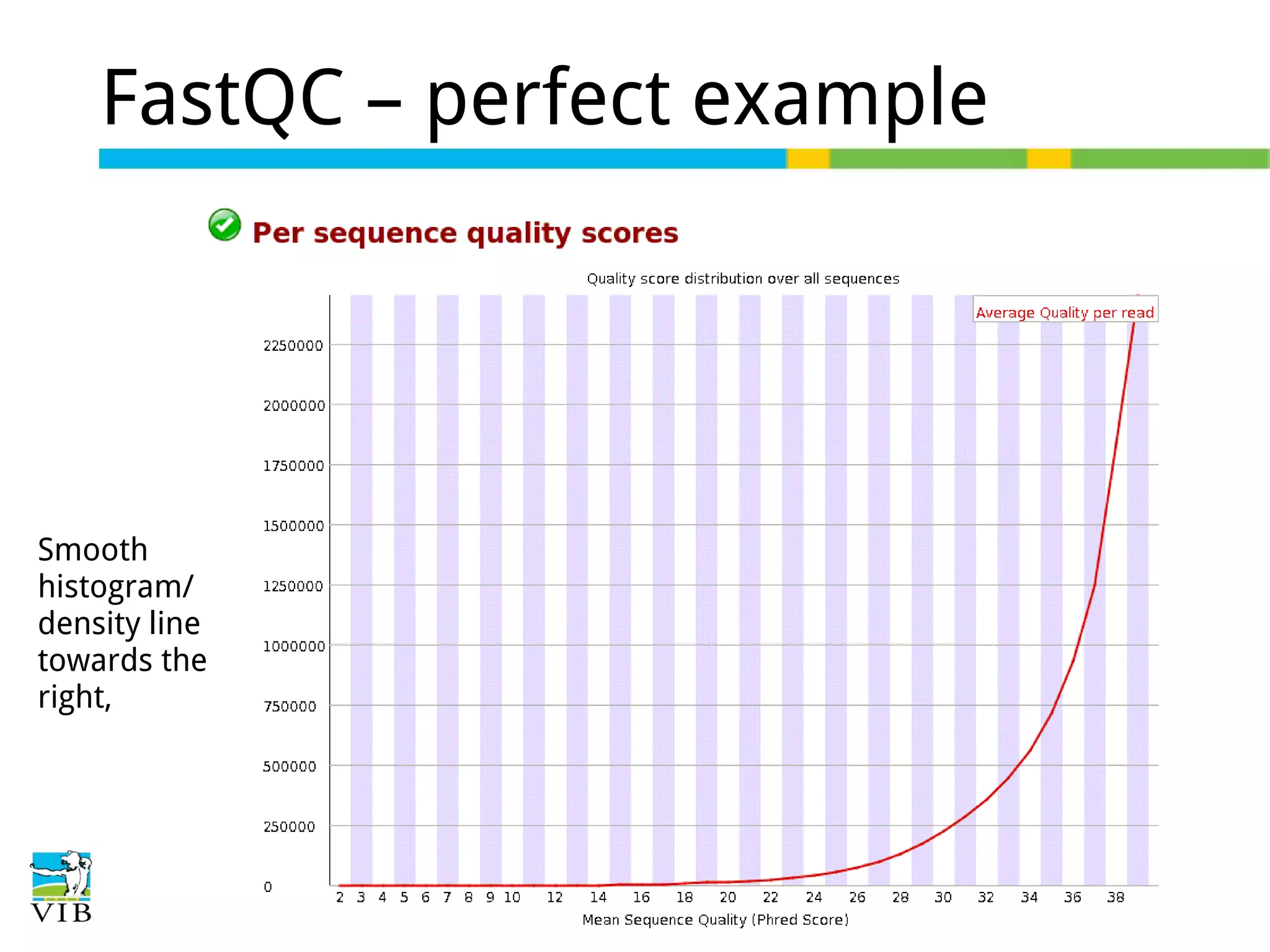
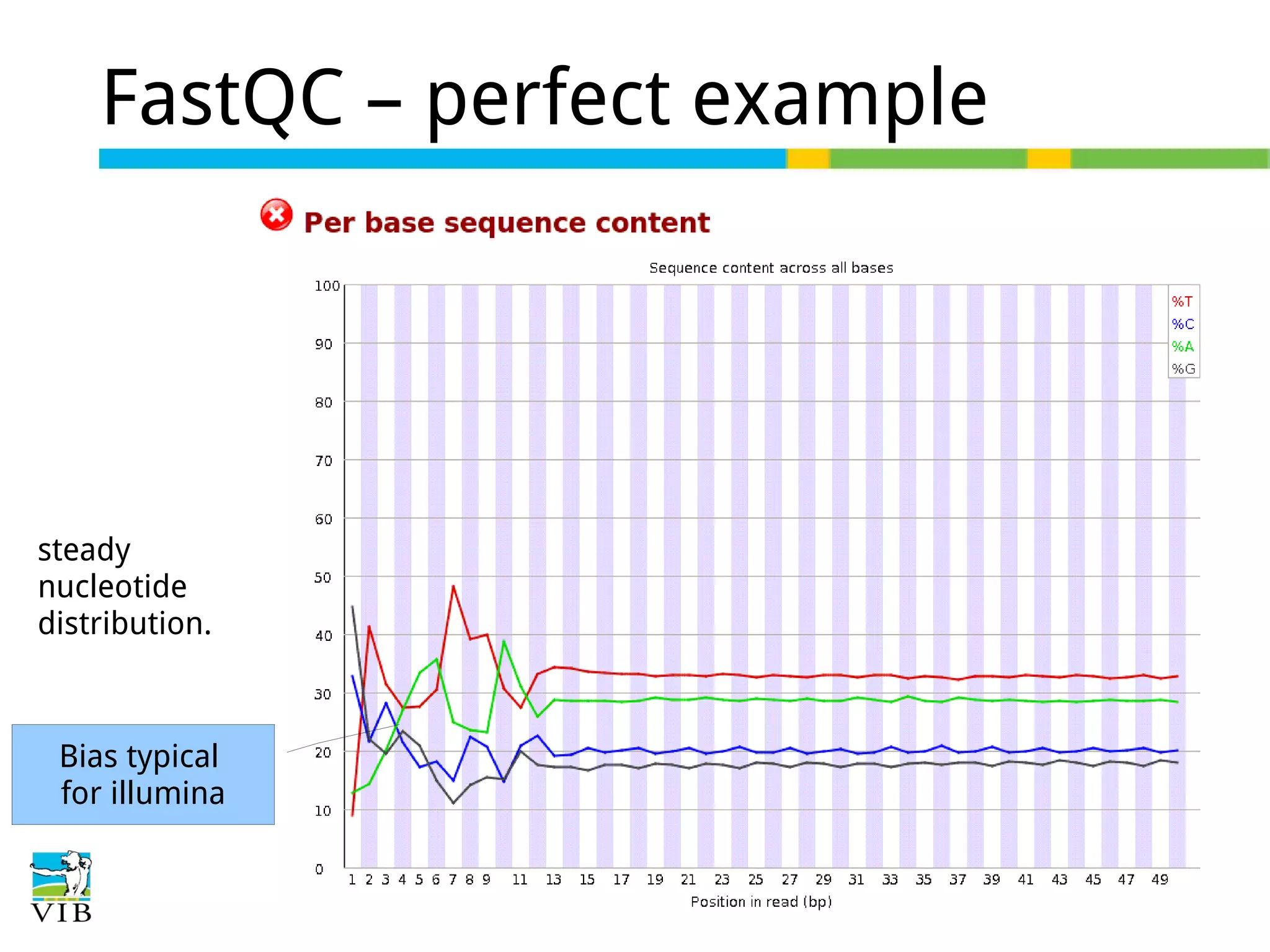
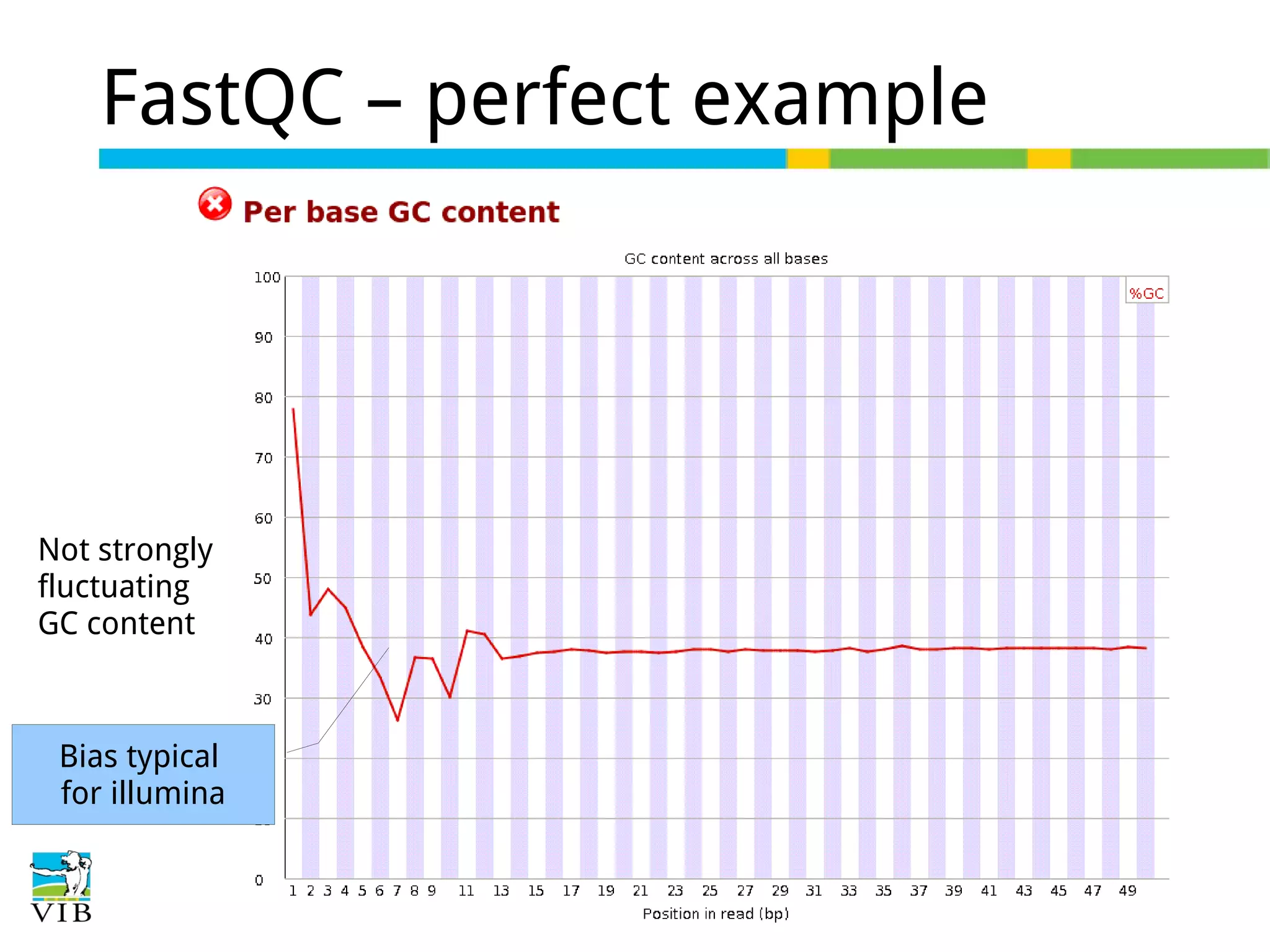
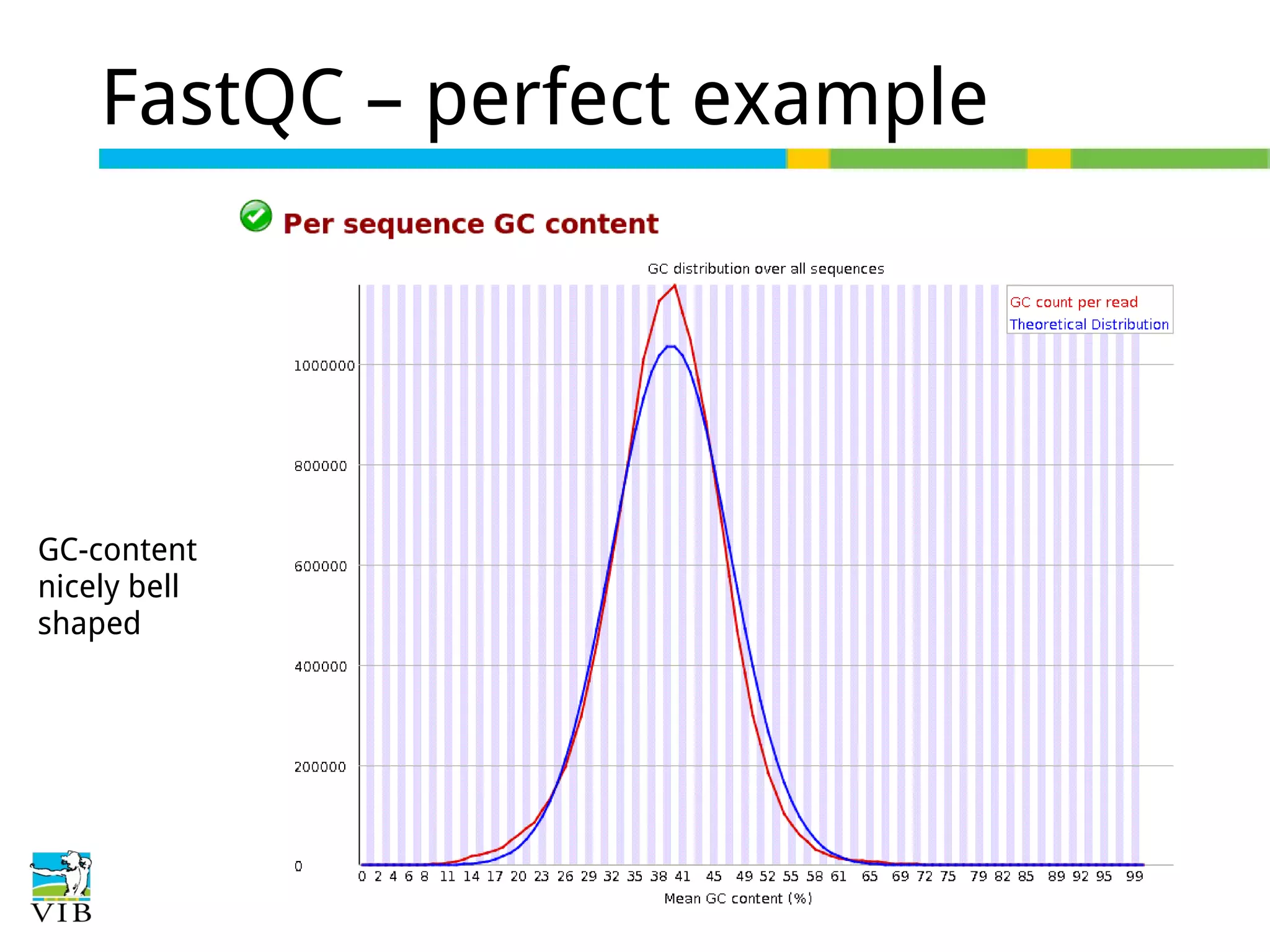
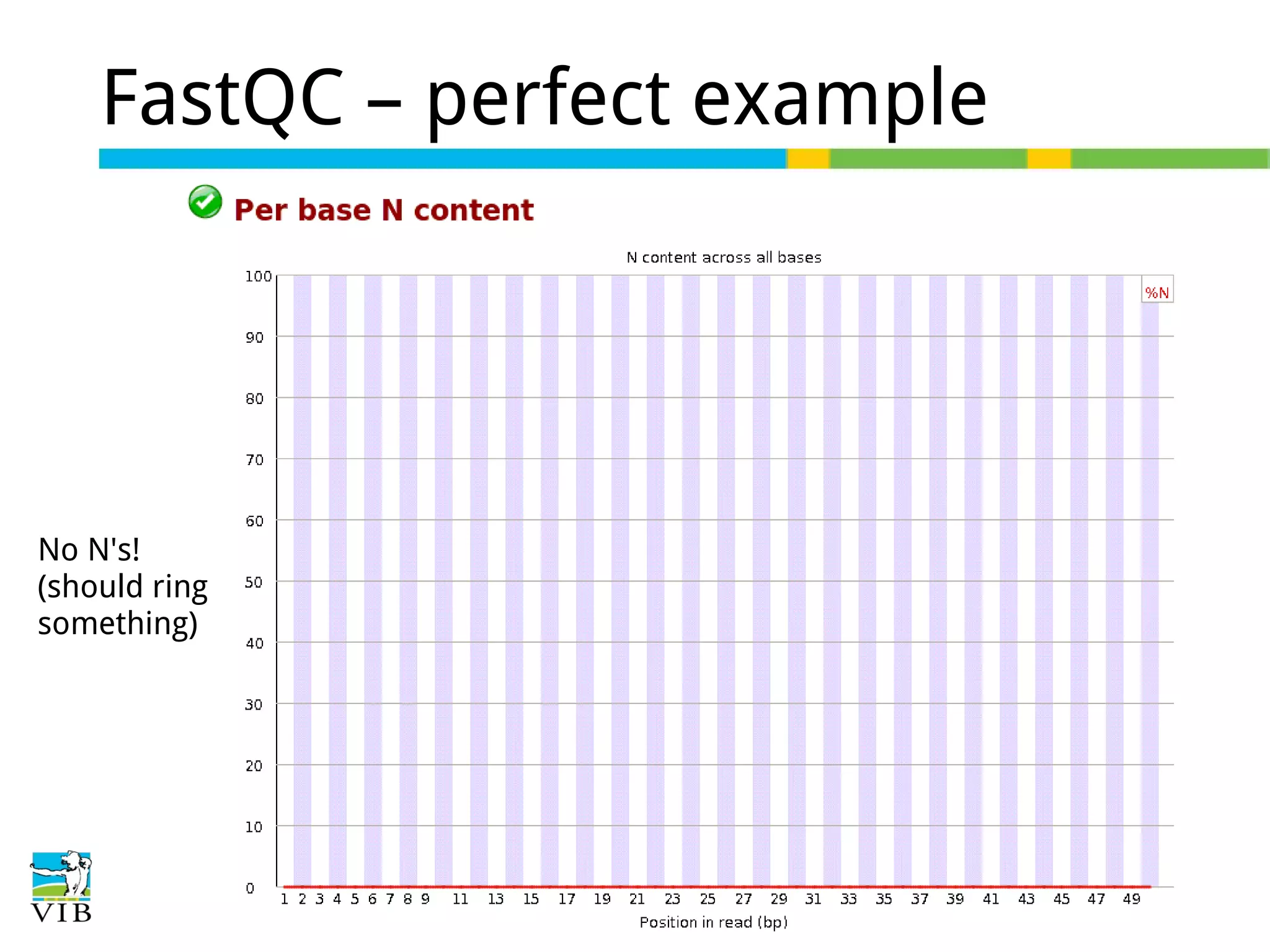
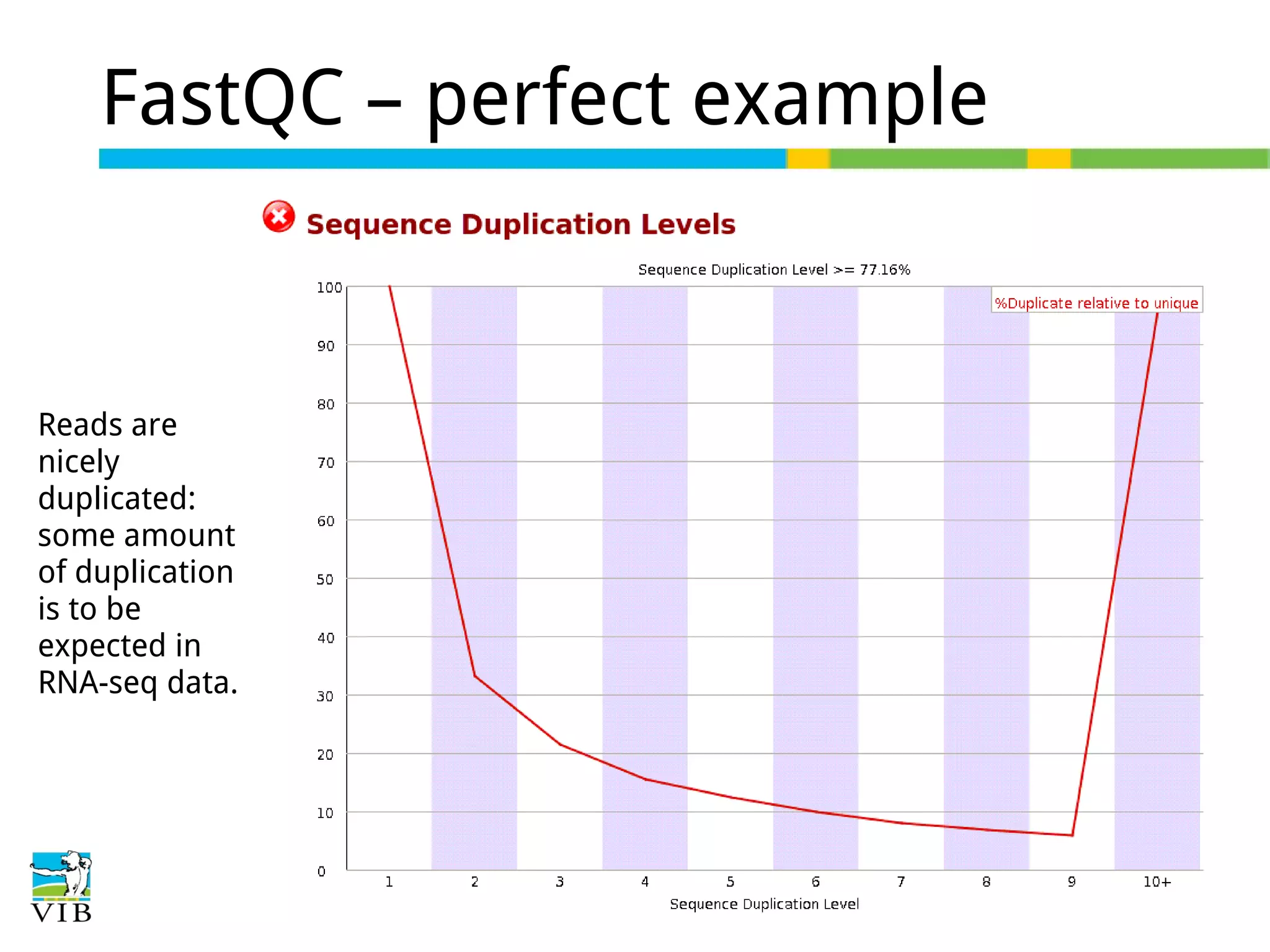
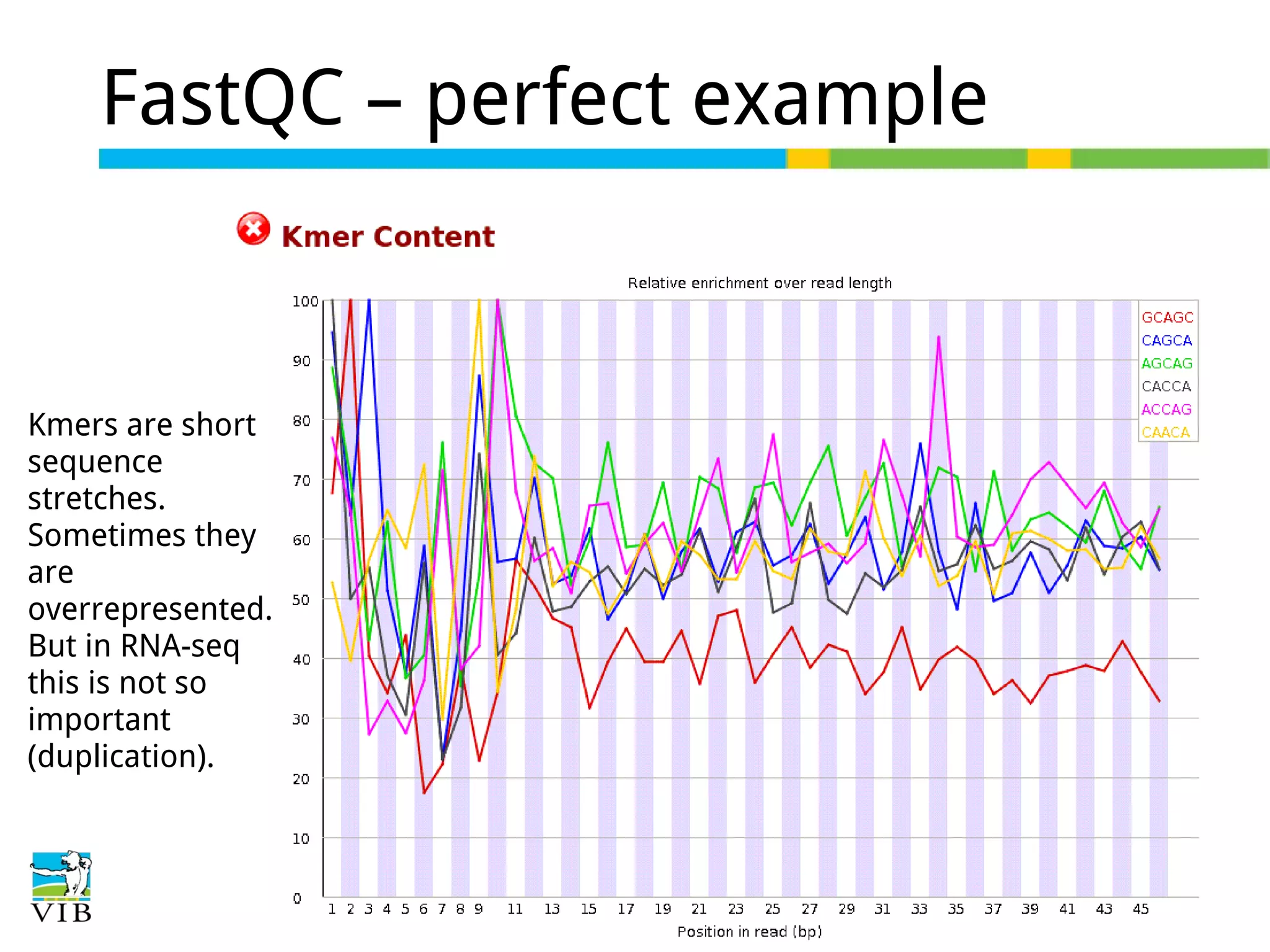
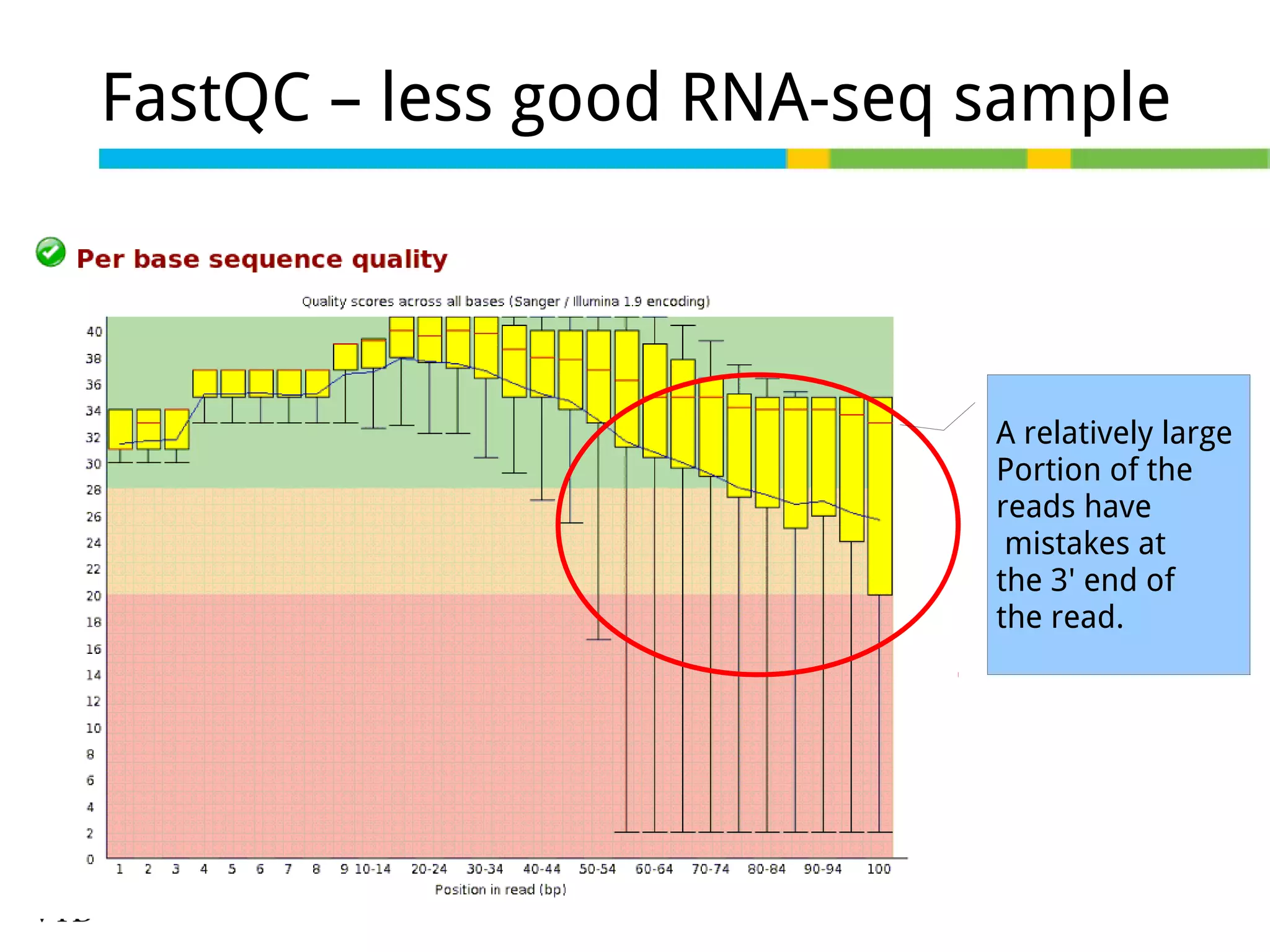
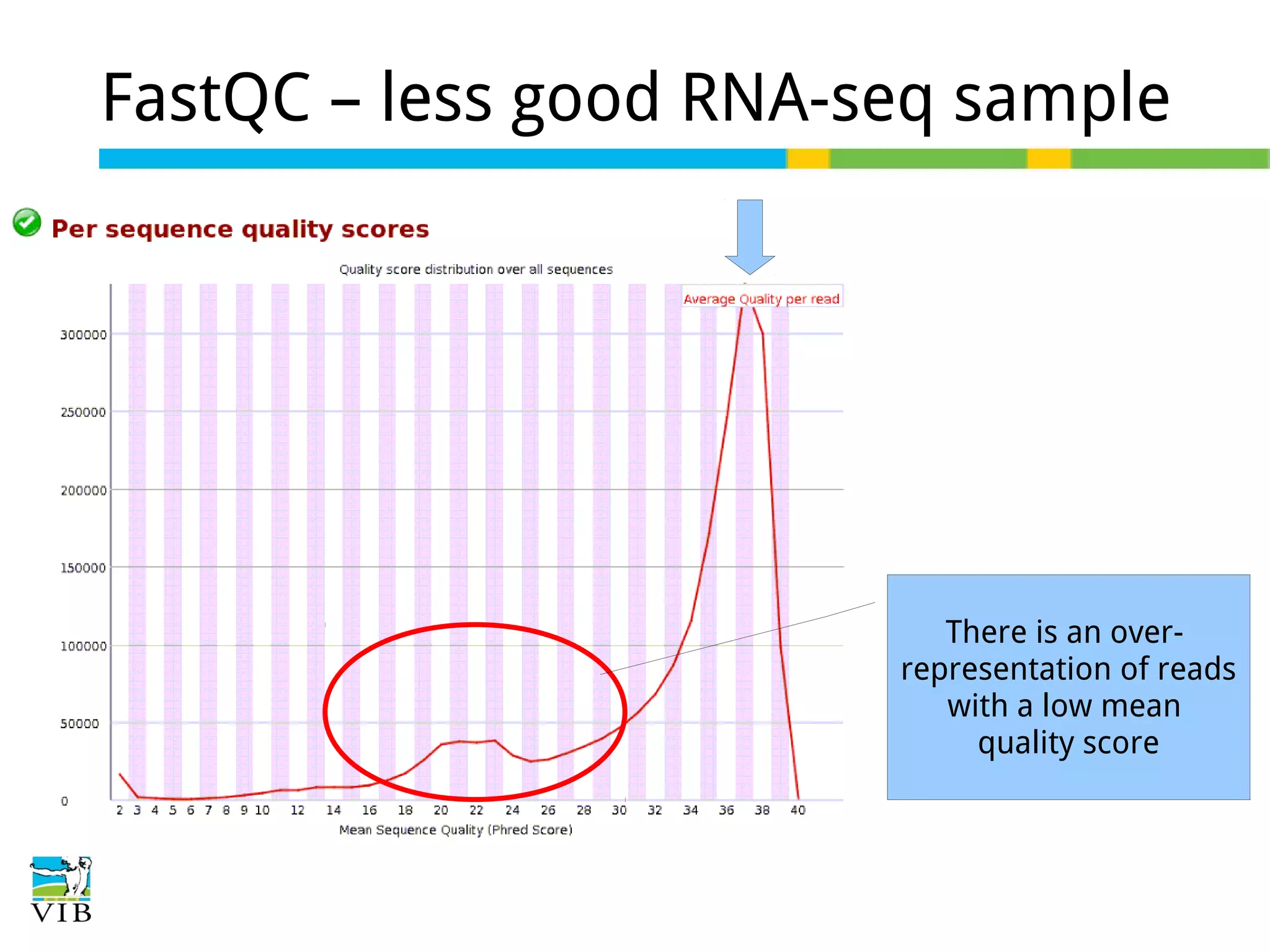
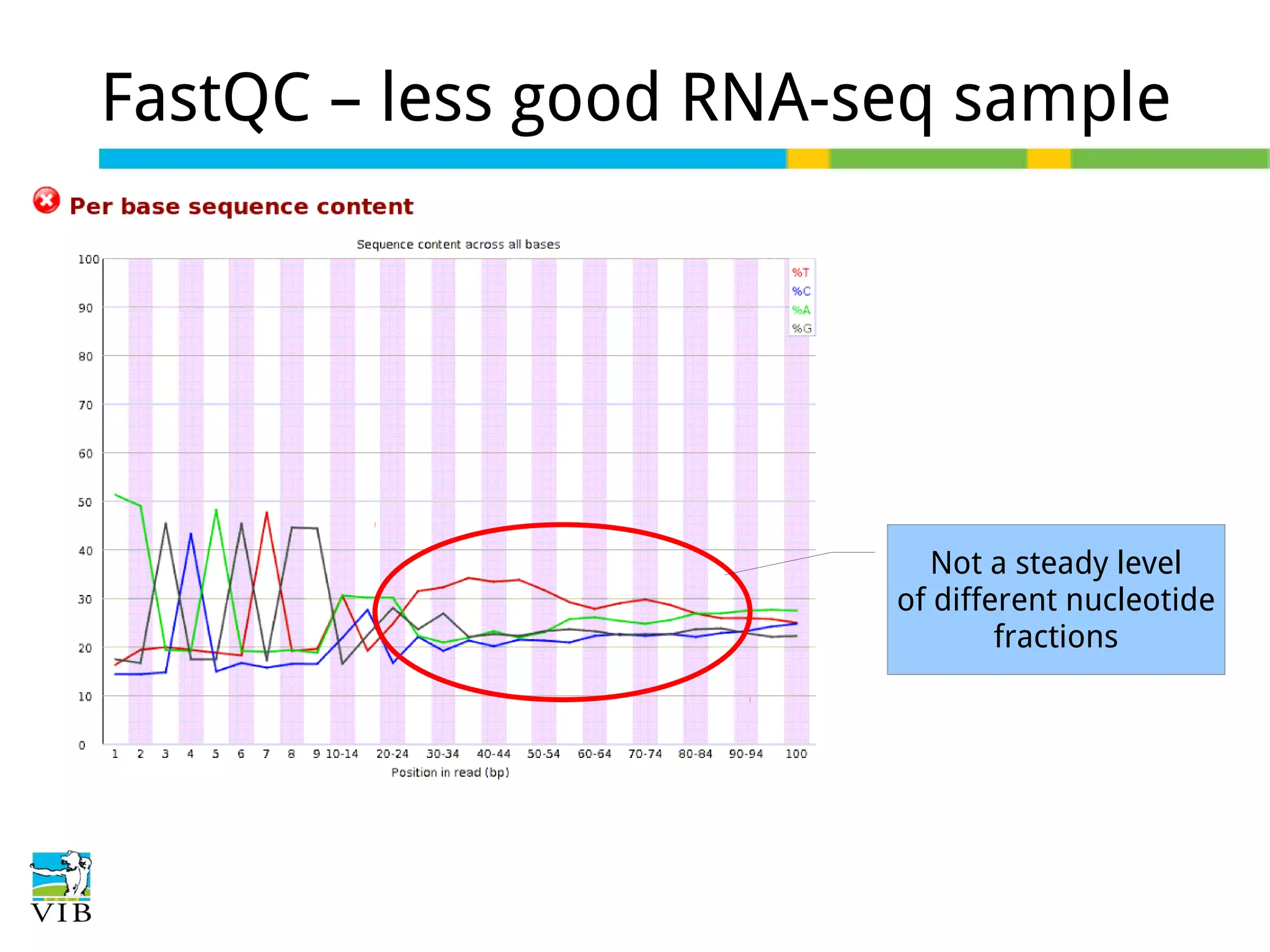
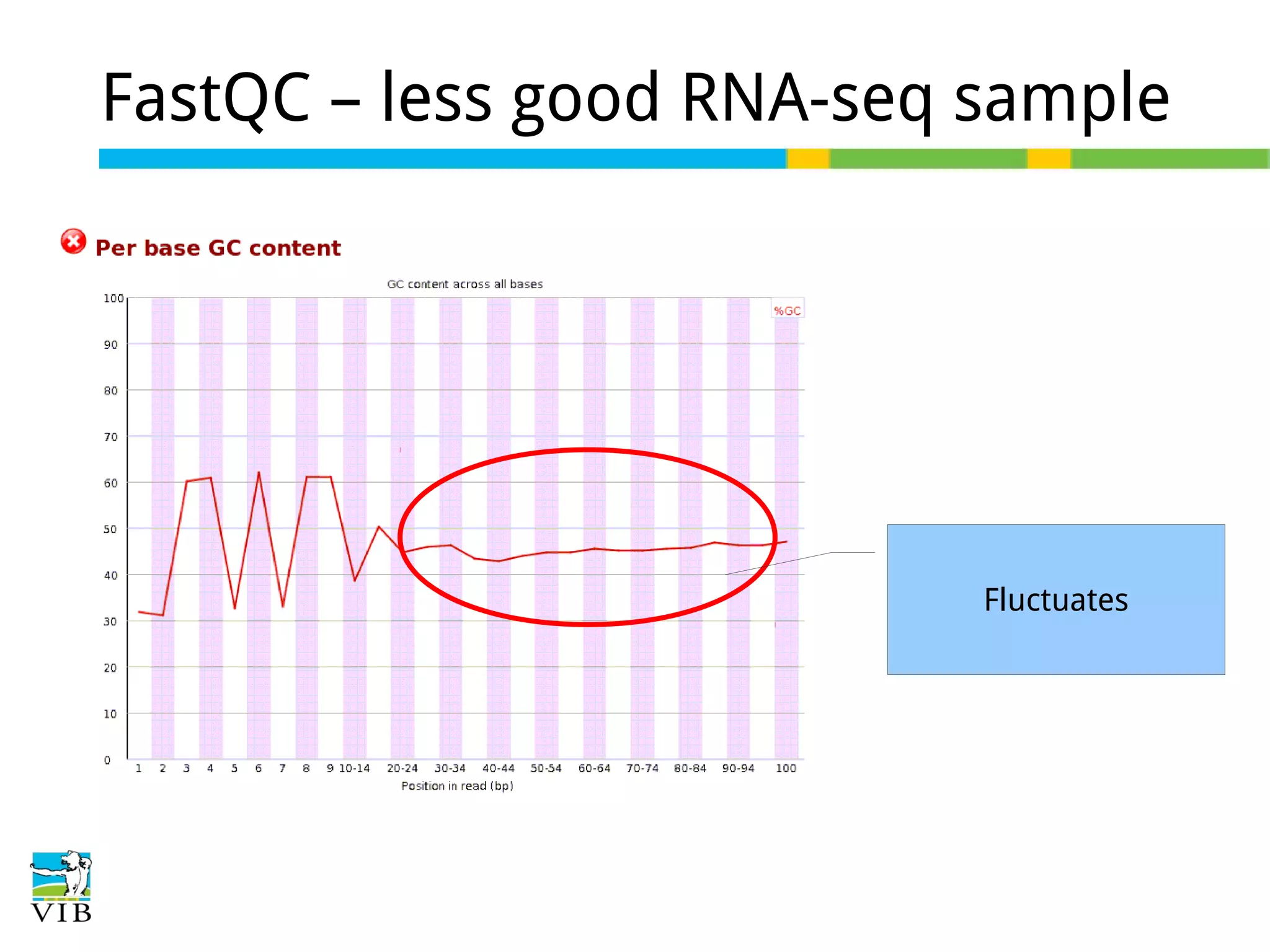
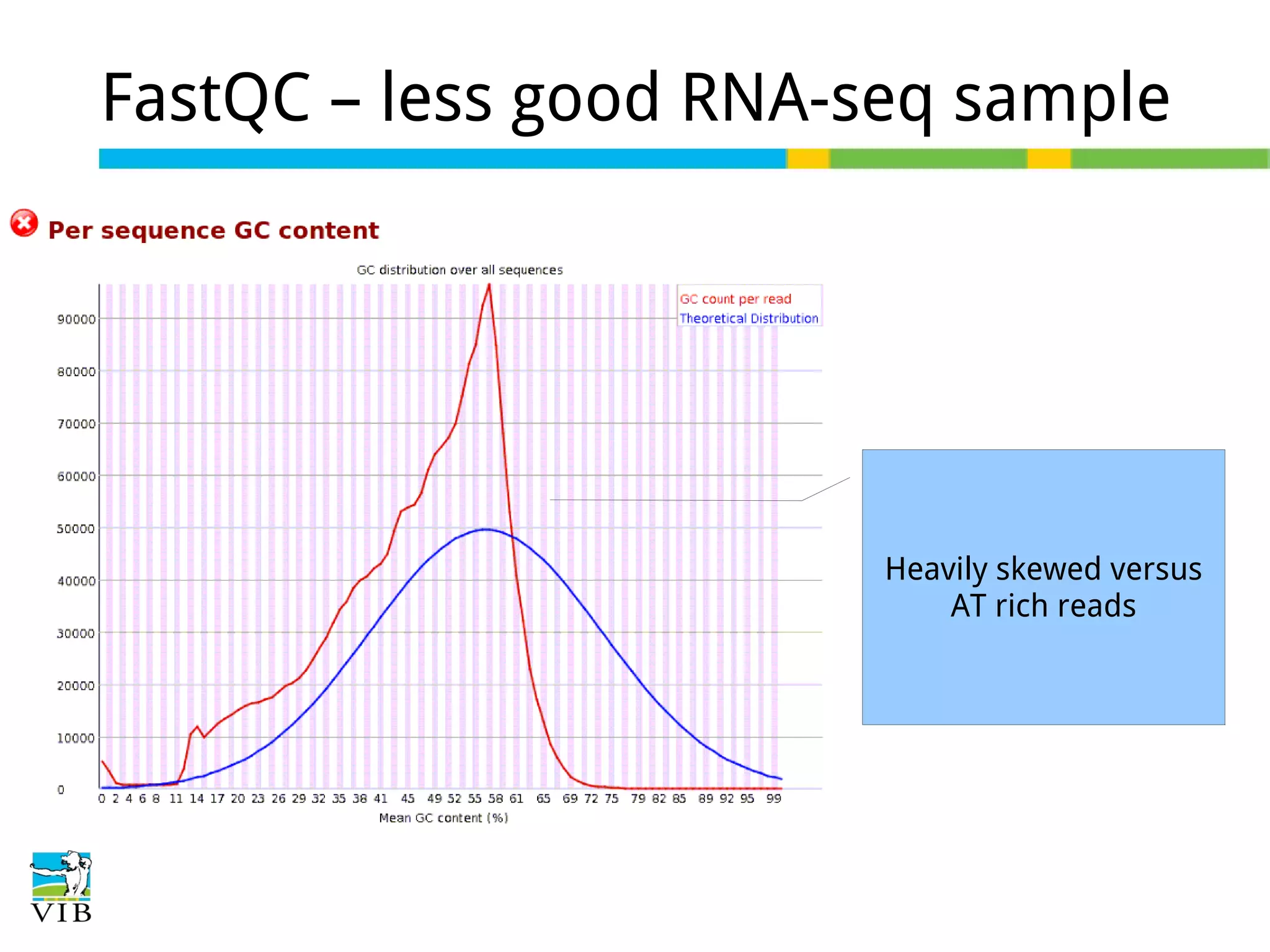
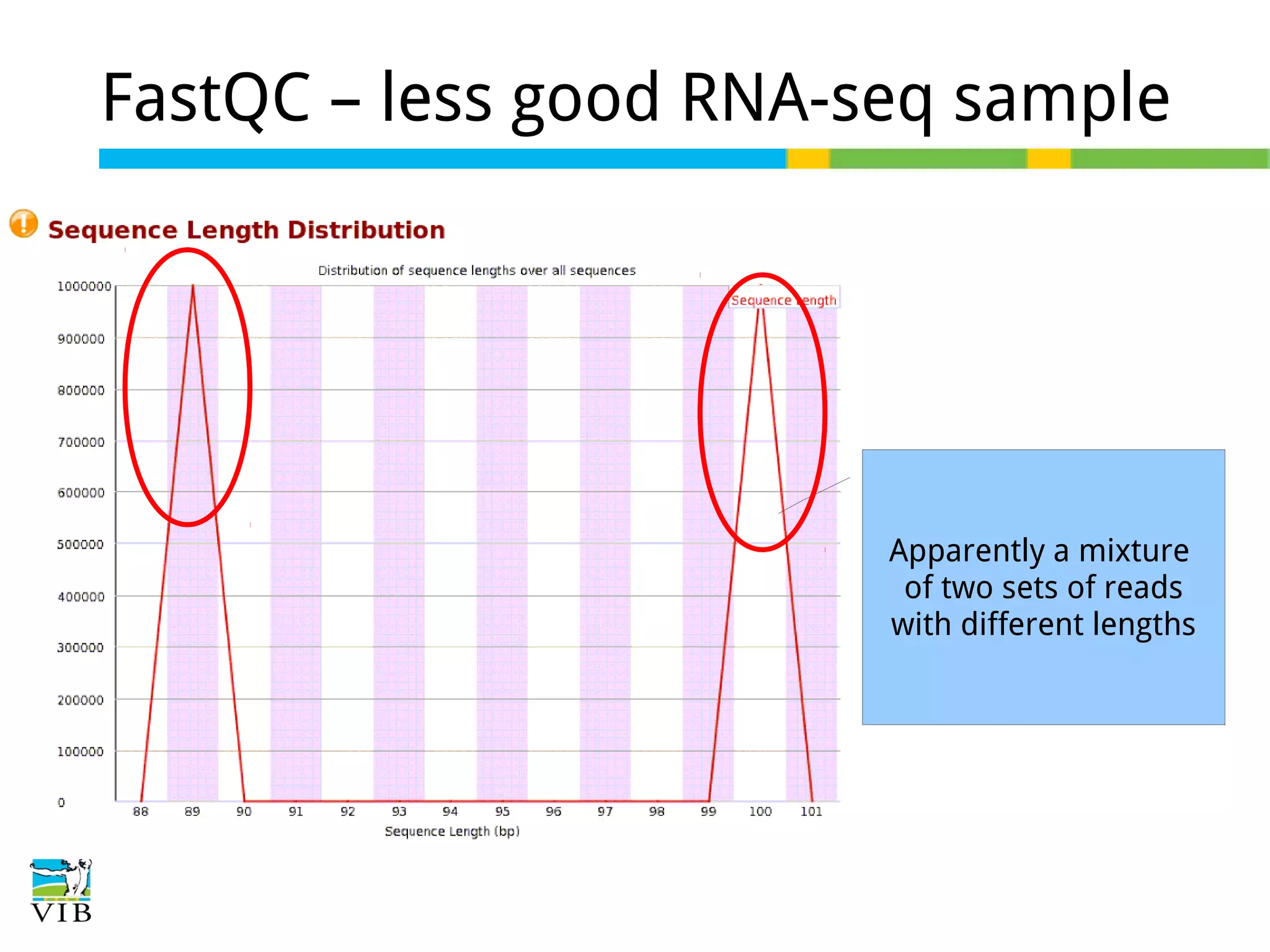
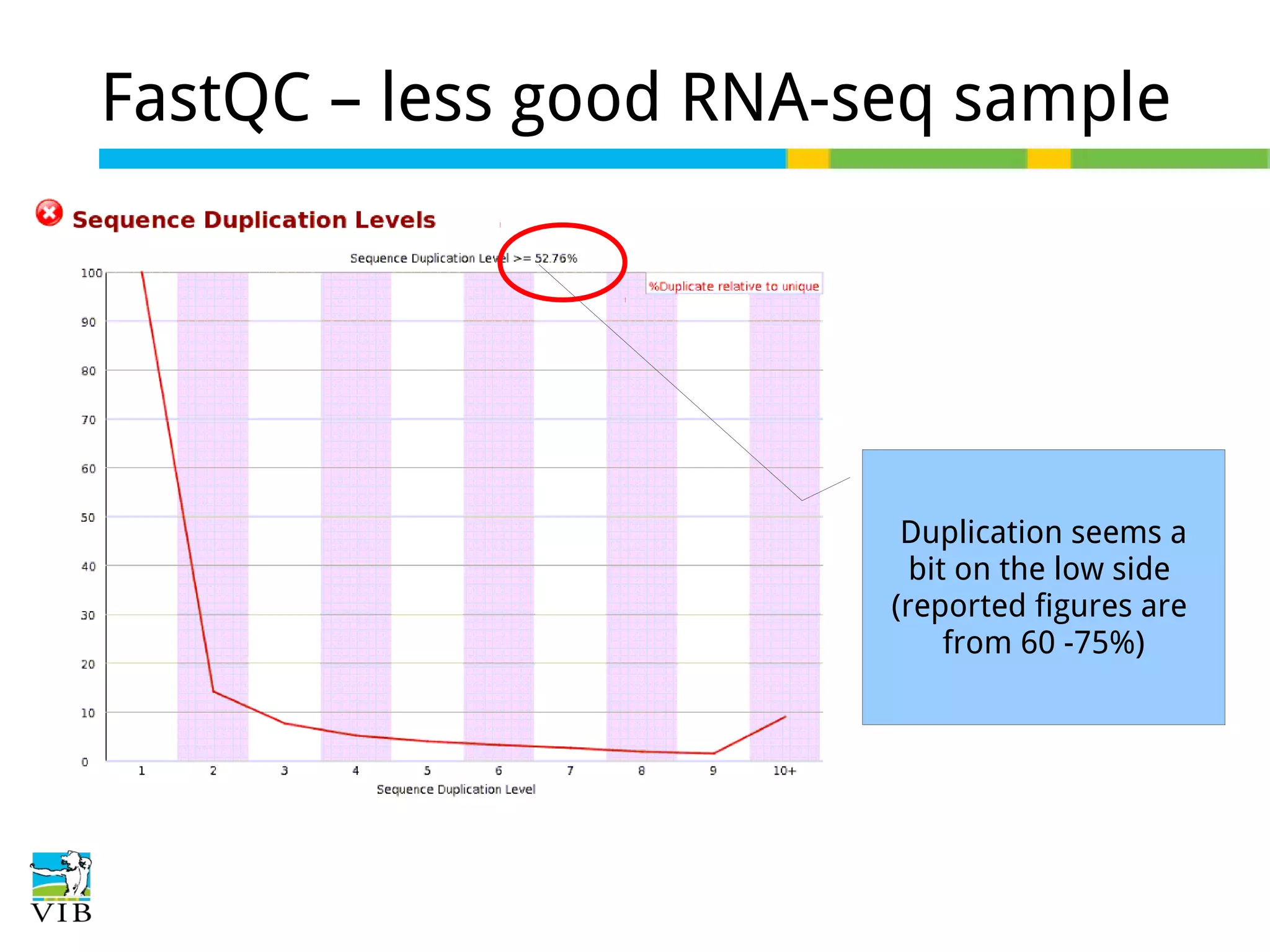
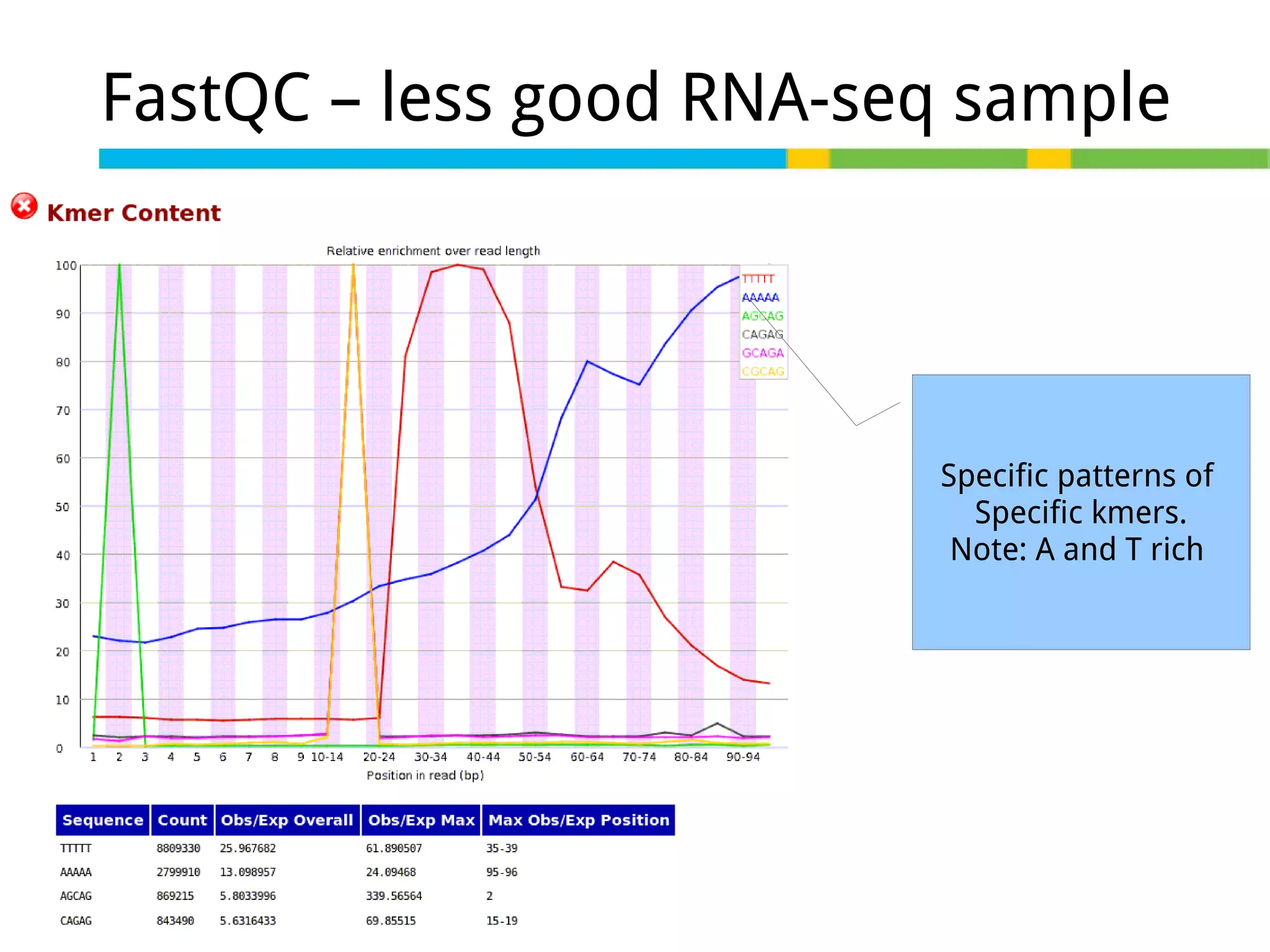
Initial steps for raw data exploration and quality control using FastQC to identify potential data issues.
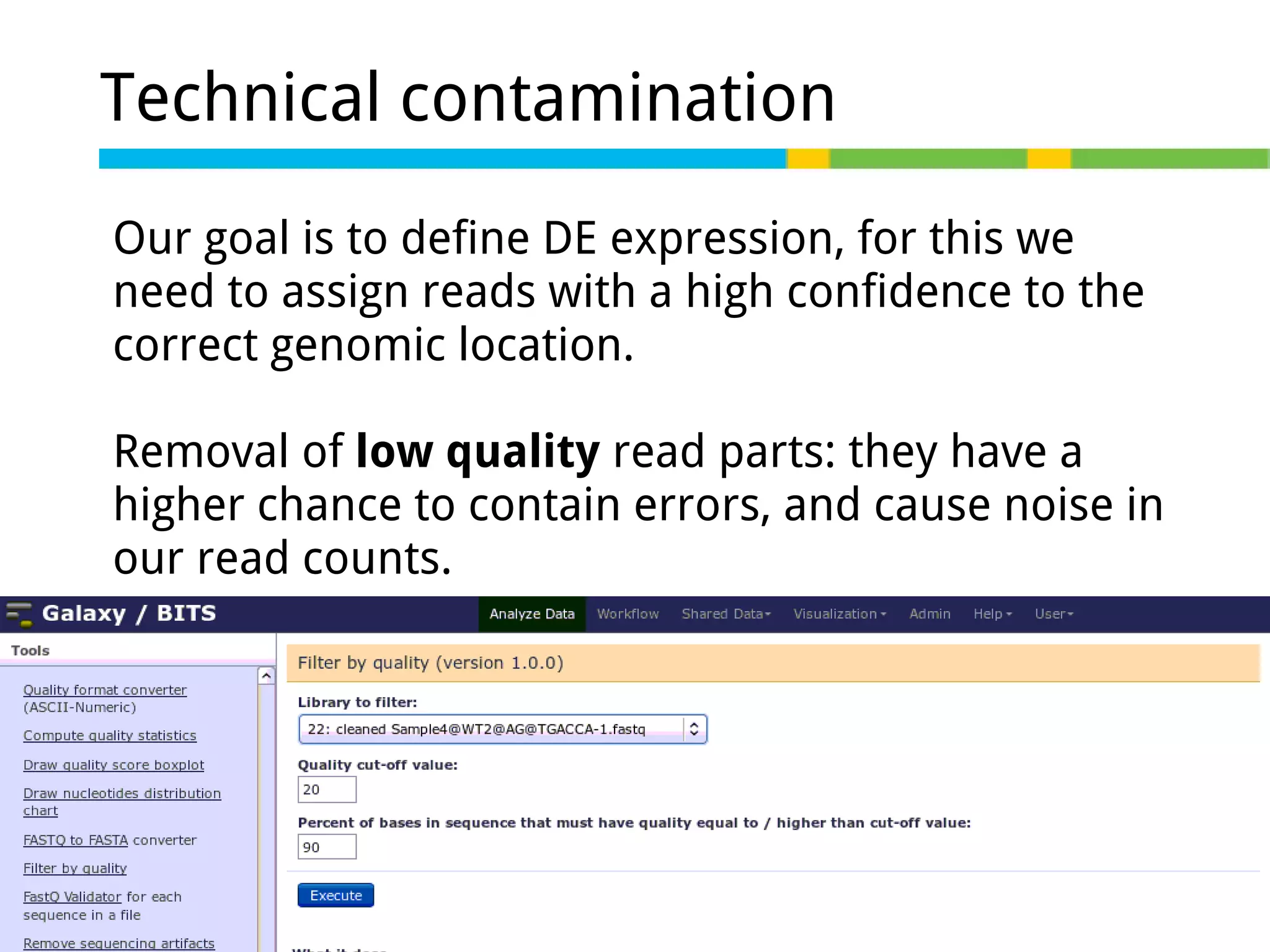
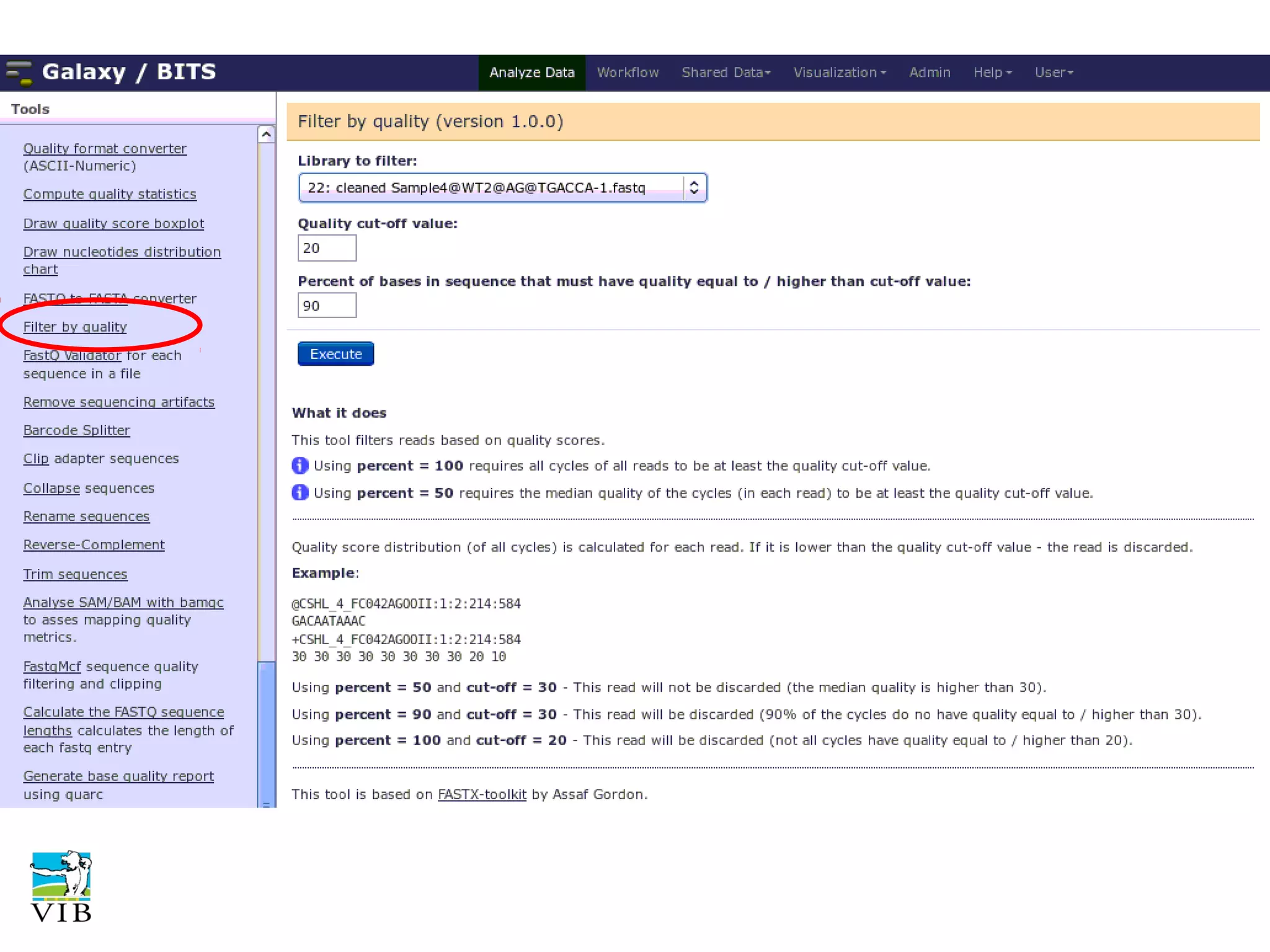
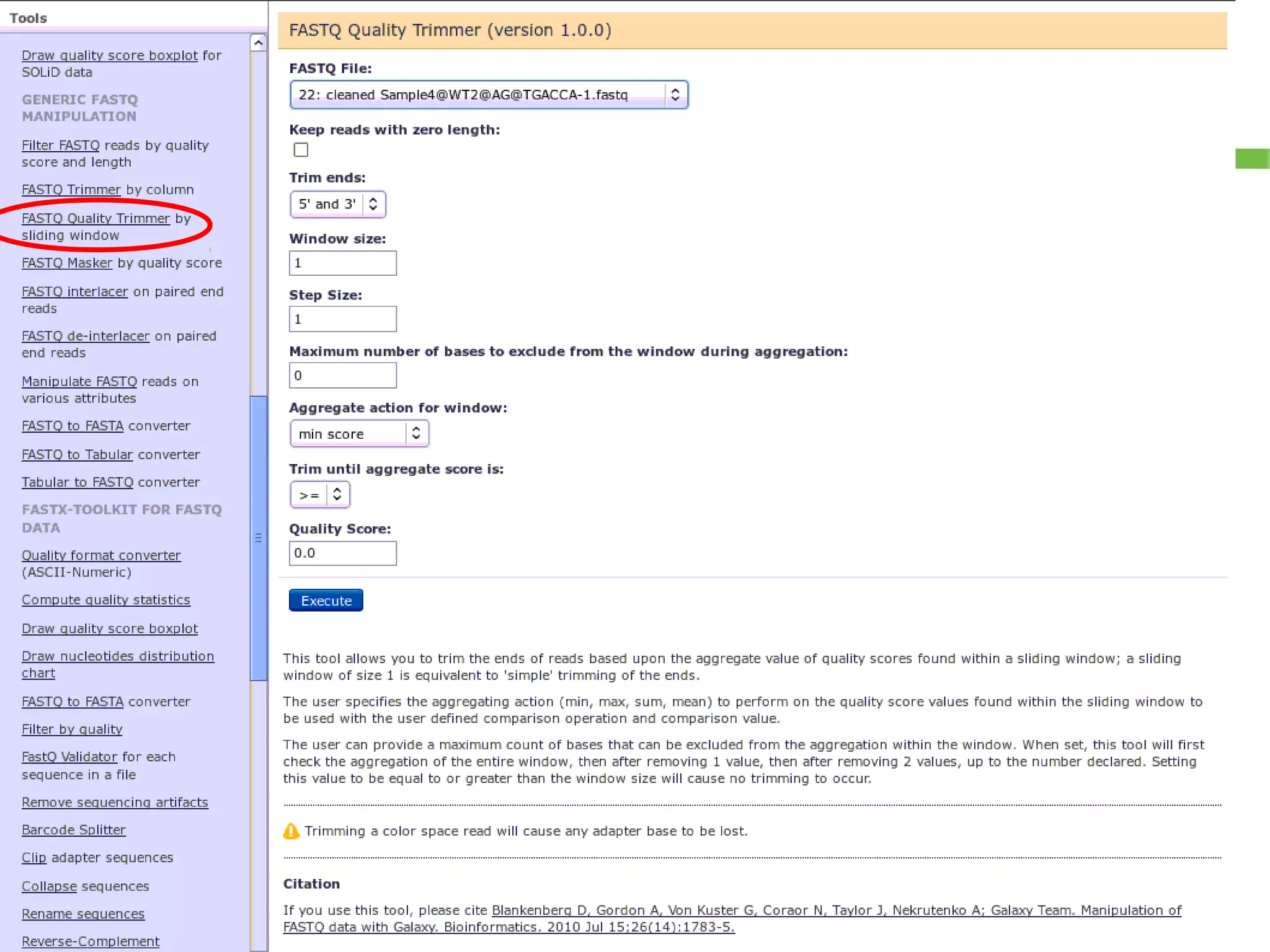
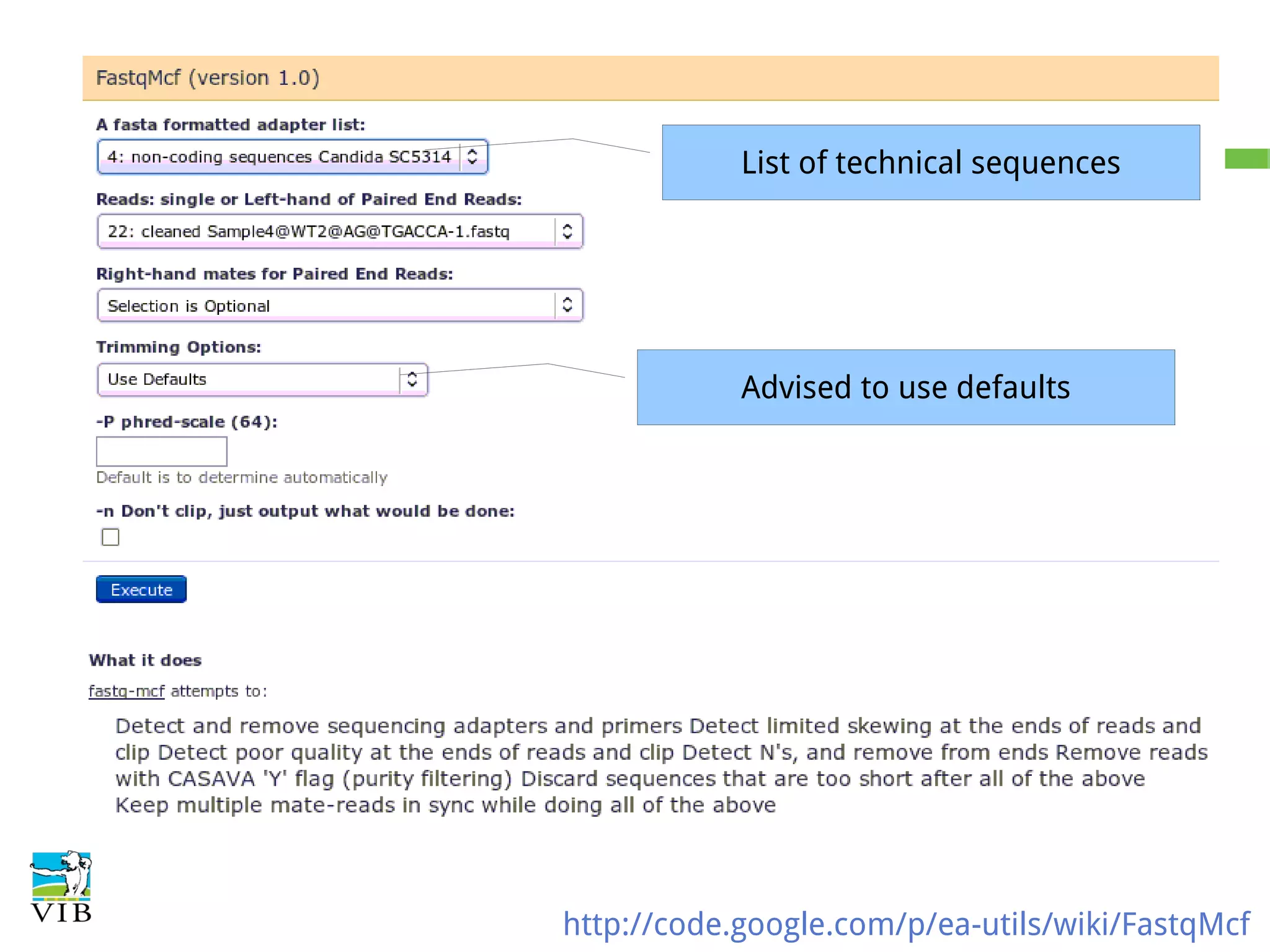
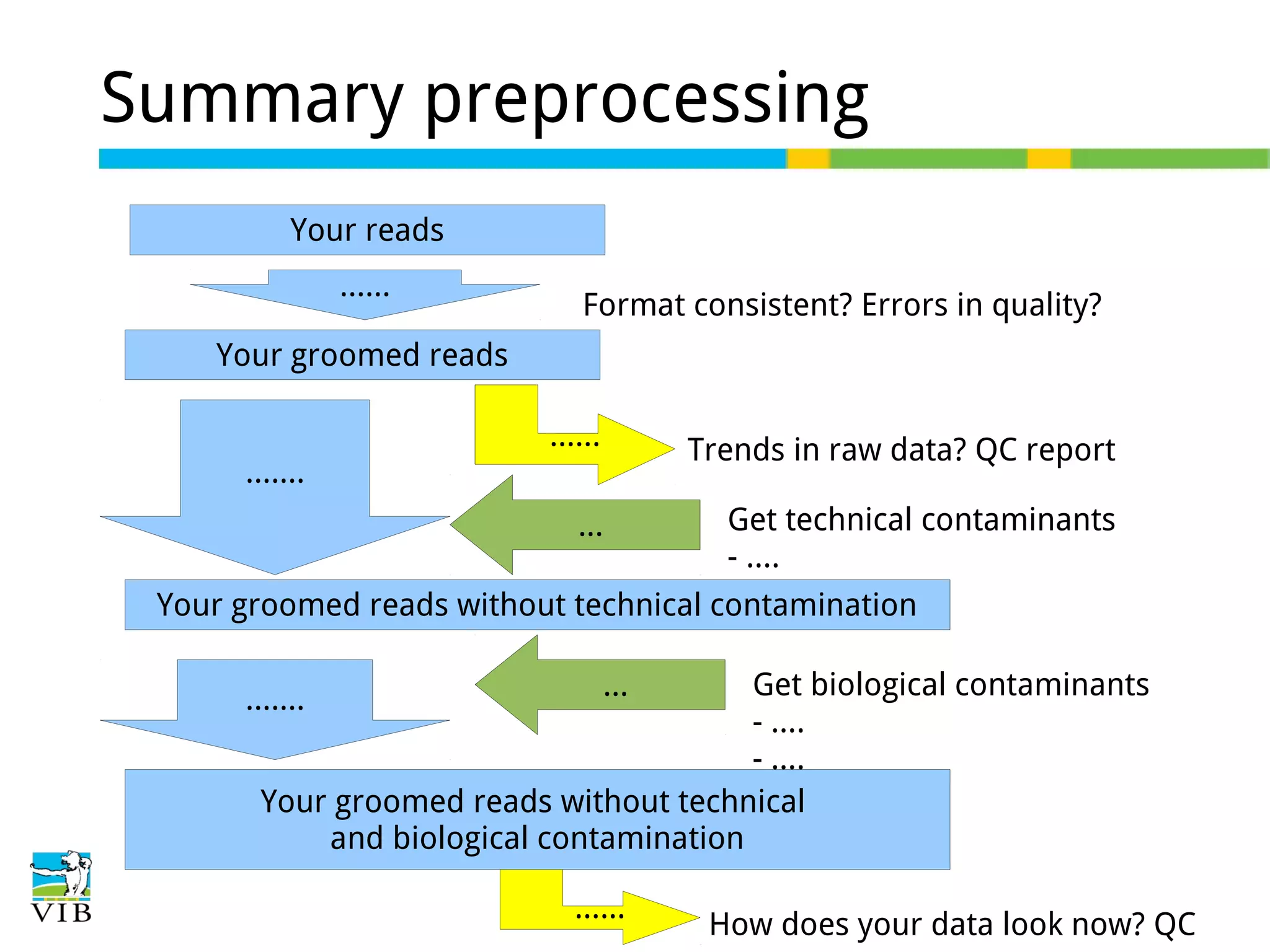
Details on preprocessing by removing unwanted sequences (technical contamination) to ensure accurate gene expression assignment.
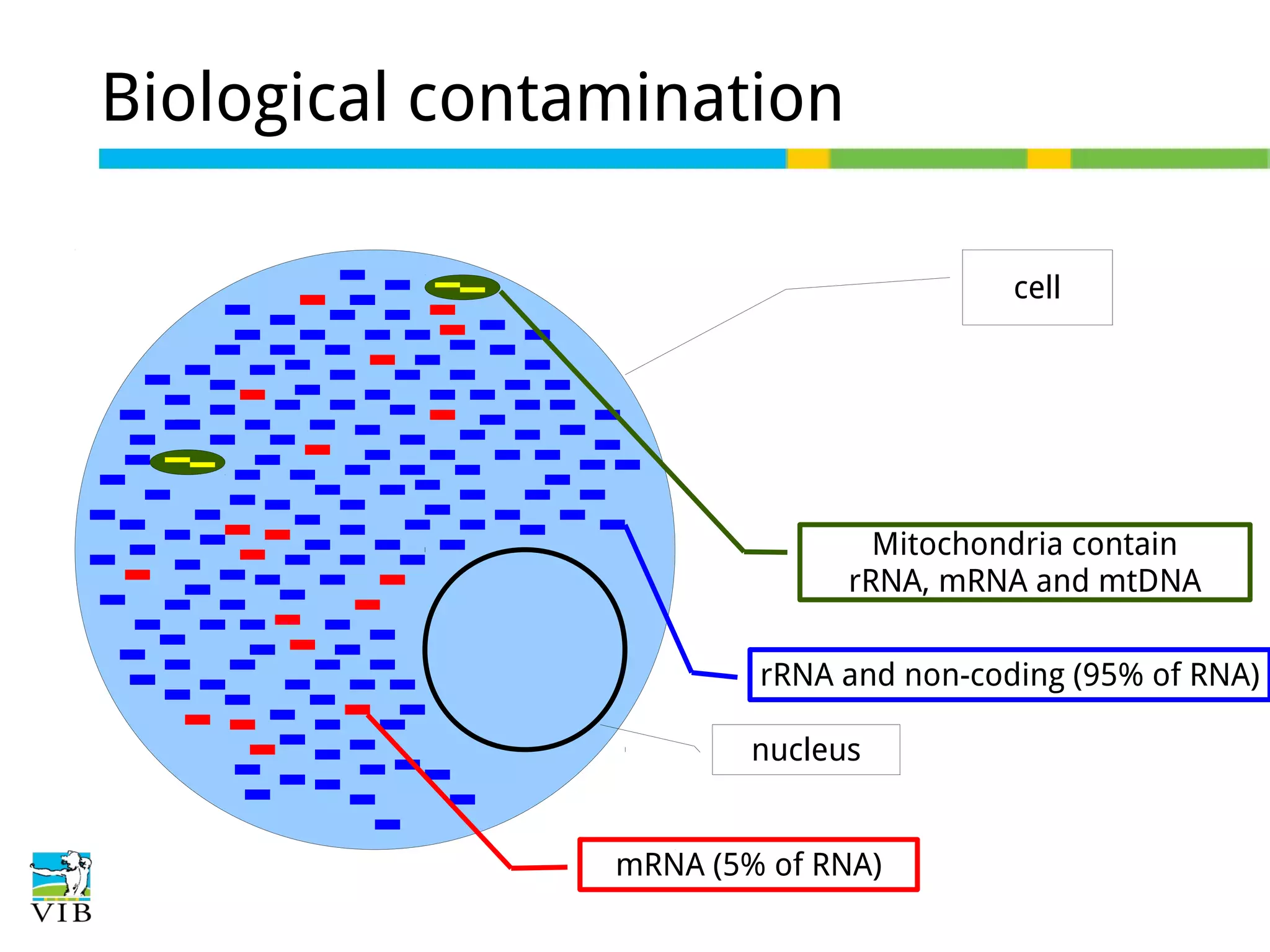
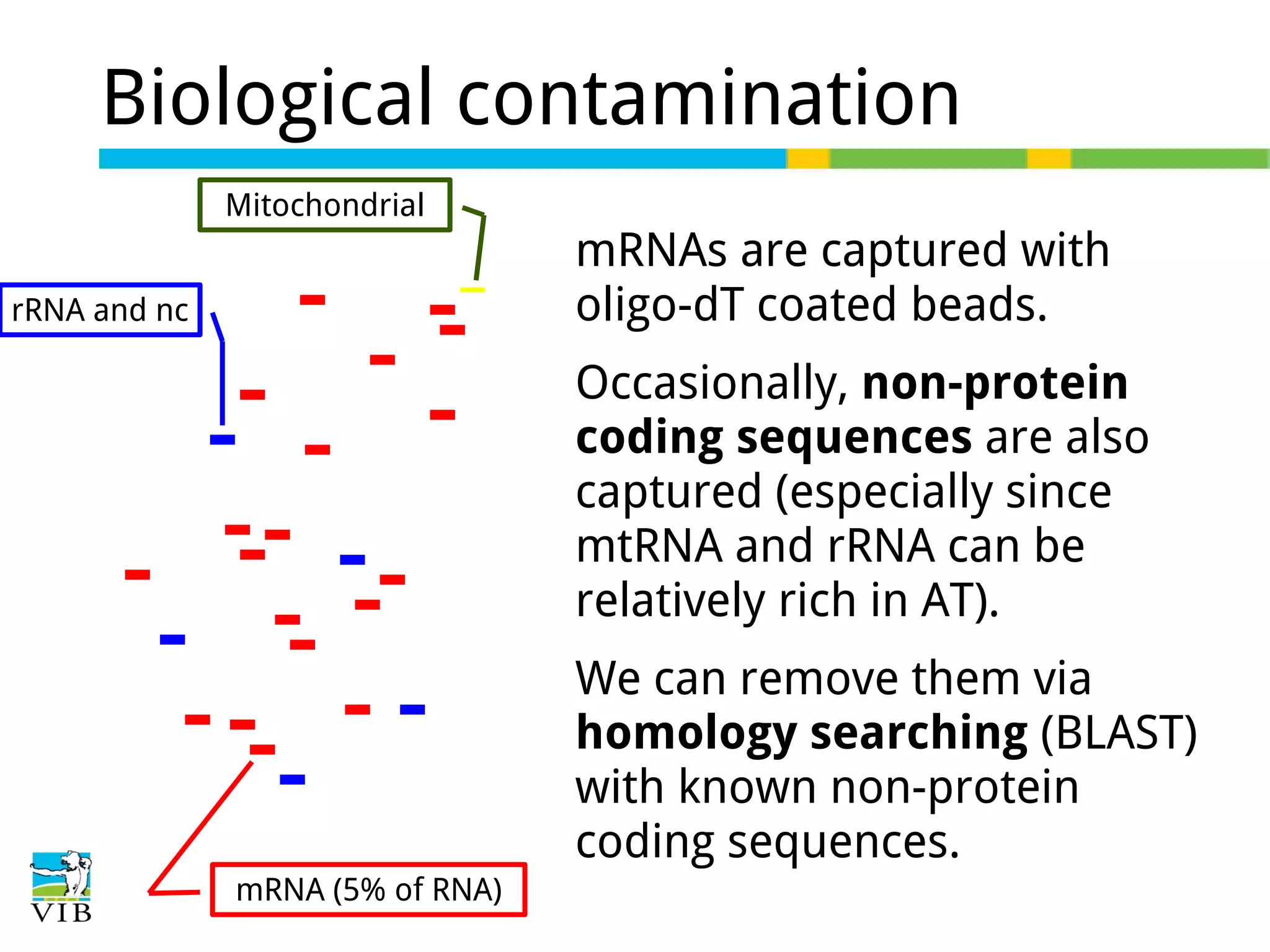
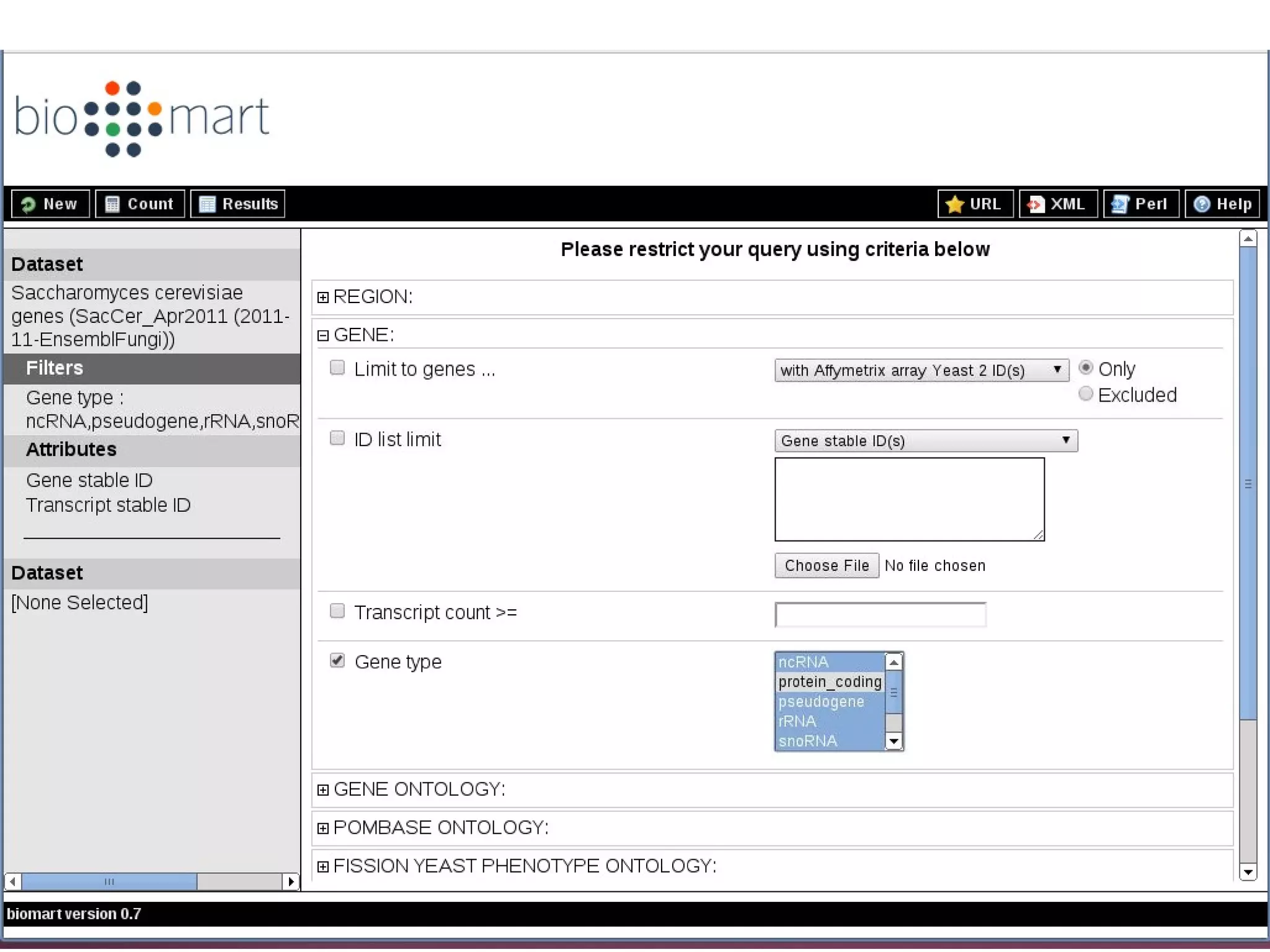
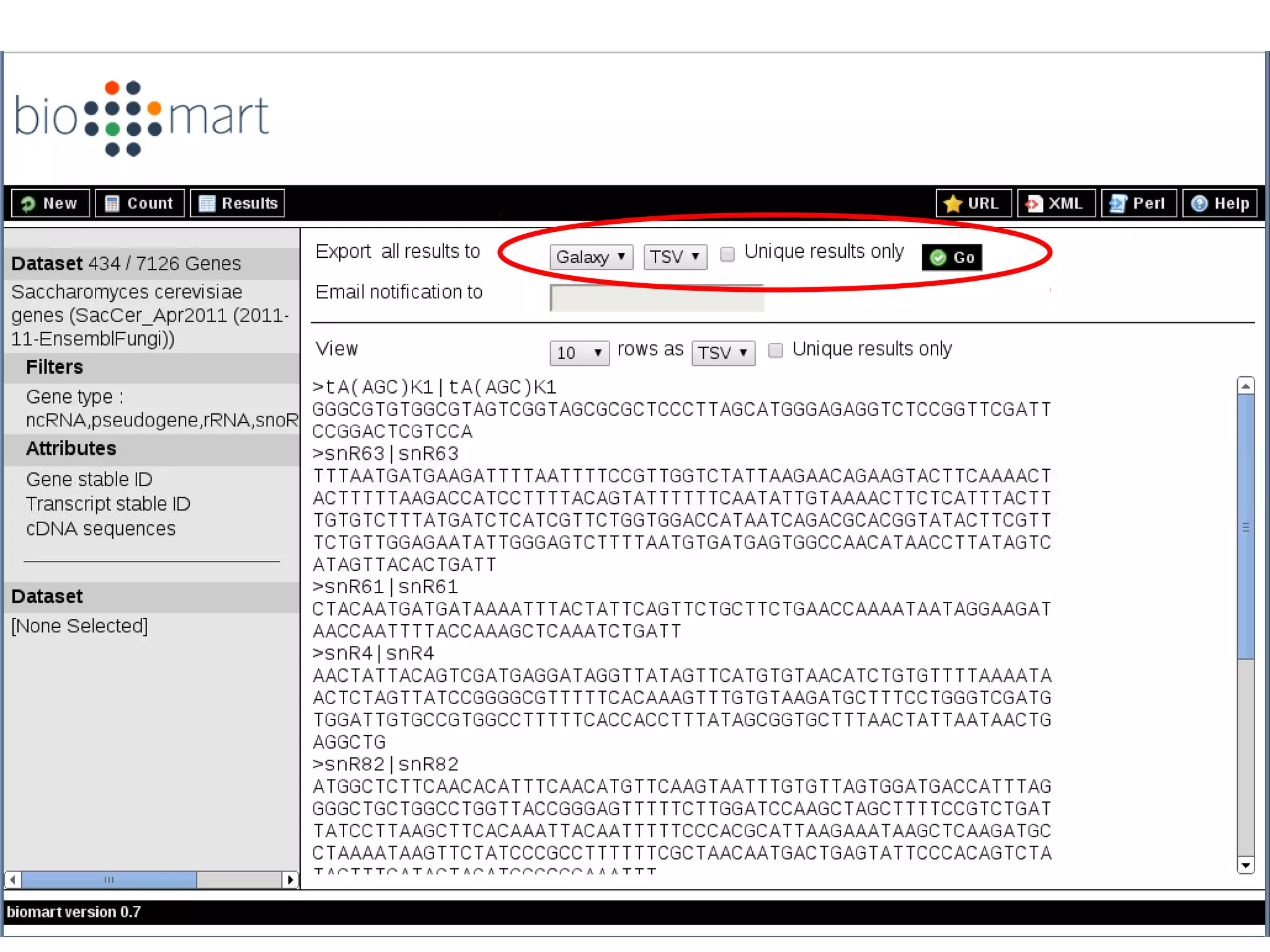
Explains biological contaminants like rRNA and mtDNA; emphasizes removal for clean data before analysis.
Workflow for preprocessing RNA-seq data in Galaxy, covering technical and biological contamination removal.
Keywords related to RNA-seq data, and an exercise on investigating and preprocessing raw data.
Short intermission in the presentation.






































































































