Porfiria
•Download as PPTX, PDF•
0 likes•132 views

The document discusses porphyria, which are metabolic disorders caused by deficiencies in enzymes involved in the heme biosynthesis pathway. The main types are hepatic or erythropoietic porphyrias depending on where excess precursors accumulate. Symptoms vary between types but can include photosensitivity, skin blistering, and liver problems. Three case studies are described that involve porphyria cutanea tarda induced by drugs like tamoxifen and resulting in liver cirrhosis in rare cases. Erythropoietic protoporphyria is also summarized, which causes photosensitivity and risk of liver disease due to protoporphyrin accumulation in blood and tissues from birth.

Recommended
More Related Content
Similar to Porfiria
Similar to Porfiria (20)
Porfiria
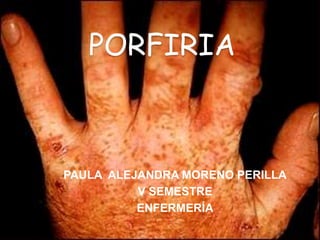
- 1. PORFIRIA PAULA ALEJANDRA MORENO PERILLA V SEMESTRE ENFERMERÍA
- 2. DEFINICIÓN Las porfirias son trastornos metabólicos y cada una de ellas es consecuencia de la deficiencia de alguna enzima específica en la vía de biosíntesis del grupo hemo. Las deficiencias enzimáticas se heredan por medio de rasgos autosómicos dominantes o recesivos, con excepción de la porfiria cutánea tardía (PCT), que suele ser esporádicas. Las porfirias se clasifican en hepáticas o eritropoyéticas, según el sitio primario de producción excesiva y acumulación de sus precursores respectivos porfirínicos o porfirinas, aunque algunas comparten características comunes. http://biblioteca.fucsalud.edu.co:2313/content.aspx?aID=4500581&searchStr=trastorno+del+metabolismo+de+las+porfirinas#4500581
- 4. MANIFESTACIONES Las principales manifestaciones de las porfirias de tipo hepático agudas son neurológicas y comprenden dolor abdominal neuropático, neuropatía motora periférica y perturbaciones psíquicas y sus ataques suelen ser desencadenados por hormonas esteroides, algunos fármacos e influencias nutricionales. Las porfirias de origen hepático generan síntomas principalmente en los adultos, pero se han identificado variantes homocigotas raras de las porfirias autonómicas dominantes de tipo hepático que por lo general surgen clínicamente antes de la pubertad.
- 6. http://www.jano.es/ficheros/images/273/273v05n02/grande/273v05n02-13134884fig01.jpg PRIMER ARTÍCULO
- 7. 1. ABSTRACT Porphyriacutaneatarda (PCT) is a metabolic disorder that results in a decrease in uroporphyrinogendecarboxylase activity. It is characterized by photosensitivity, bullae formation, and skin pigmentation. There are four types of PCT: acquired, familial, toxic, and hepatoerythropoietic. Uroporphyrin levels are elevated in the urine of PCT patients.
- 8. PCT can be differentiated from other porphyrias by its clinical characteristics and the porphyrin levels in the serum, erythrocytes, urine, and feces. This metabolic disorder can lead to liver dysfunction as well as histological changes such as fatty infiltration or hepatic fibrosis. PCT rarely manifests as liver cirrhosis. We report herein a case of PCT-induced liver cirrhosis that progressed to hepatic failure.
- 9. NOMBRE DEL ARTÍCULO Liver cirrhosis induced by porphyriacutaneatarda: a case report and review. AUTORES: KwangGyun Lee,*JongJinHyun,*YeonSeok Seo,* Bora Keum,*HyungJoonYim,*Yoon Tae Jeen,* Hong Sik Lee,*Hoon Jai Chun,* Chang Duck Kim,* Ho SangRyu,* and Soon Ho Um*† LINK:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3021615/?tool=pubmed
- 12. 2. ABSTRACT Porphyriacutaneatarda (PCT) results from a decrease in the activity of uroporphyrinogendecarboxylase. In the sporadic form, the decrease in the activity is restricted to the liver and is generally related to alcohol, estrogens, iron overload, hepatitis C infection, and halogenated aromatic hydrocarbons. We describe the development of porphyriacutaneatarda in a 53-year-old woman one year after breast cancer surgery and the initiation of treatment with tamoxifen.
- 13. No additional drugs were prescribed. After tamoxifen was discontinued, a gradual clinical and laboratorial improvement was noticed suggesting a causative role of the drug. There are many reports discussing tamoxifen side-effects, but there are only three case reports in the literature that describe tamoxifen as a probable trigger of porphyriacutaneatarda. In this report, the potential porphyrinogenicity of tamoxifen and clinical implications are the targets of our discussion.
- 14. NOMBRE DEL ARTÍCULO: Porphyriacutanea tarda inducedbytamoxifen AUTORES: MariaJ Cruz MD, Sergio Alves MD, Teresa Baudrier MD, Filomena AzevedoMD LINK:http://dermatology.cdlib.org/1609/1_case_reports/2_10-00103/cruz.html
- 17. 3. ABSTRACT Erythropoieticprotoporphyria (EPP) is an inherited disorder of the haem metabolic pathway characterised by accumulation of protoporphyrin in blood, erythrocytes and tissues, and cutaneous manifestations of photosensitivity. EPP has been reported worldwide, with prevalence between 1:75,000 and 1:200,000. It usually manifests in early infancy upon the first sun exposures. EPP is characterised by cutaneous manifestations of acute painful photosensitivity with erythema and oedema, sometimes with petechiae, together with stinging and burning sensations upon exposure to sunlight, without blisters.
- 18. These episodes have a variable severity depending on the exposure duration and may result in chronic permanent lesions on exposed skin. As protoporphyrin is a lipophilic molecule that is excreted by the liver, EPP patients are at risk of cholelithiasis with obstructive episodes, and chronic liver disease that might evolve to rapid acute liver failure.
- 19. In most patients, EPP results from a partial deficiency of the last enzyme of the haem biosynthetic pathway, ferrochelatase, EC 4.99.1.1/FECH (encoded by the FECH gene). EPP appears to be inherited as an autosomal dominant disease, the clinical expression of which is modulated by the presence of the hypomorphicFECH IVS3-48C allele trans, but recessive inheritance with two mutated FECH alleles has also been described.
- 20. In about 2% of patients, overt disease was recently shown to be caused by gain-of-function mutations in the erythroid-specific aminolevulinic acid synthase 2 (ALAS2/ALAS, EC 2.3.1.27) gene and named X-linked dominant protoporphyria. Diagnosis is established by finding increased levels of protoporphyrin in plasma and red blood cells, and detection of a plasma fluorescence peak at 634 nm.
- 21. Investigations for hepatic involvement, ferrochelatase activity level, genetic analysis (FECH mutations, presence of the hypomorphicFECH IVS3-48C allele trans and ALAS2 mutations) and family studies are advisable. Differential diagnosis includes phototoxic drug reactions, hydroavacciniforme, solar urticaria, contact dermatitis, angio-oedema and, in some cases, other types of porphyria.
- 22. Management includes avoidance of exposure to light, reduction of protoporphyrin levels and prevention of progression of possible liver disease to liver failure. As the major risk in EPP patients is liver disease, a regular follow-up of hepatic involvement is essential. Sequential hepatic and bone marrow transplantation should be
- 23. considered as a suitable treatment for most severe cases of EPP with hepatic involvement. EPP is a lifelong disorder whose prognosis depends on the evolution of the hepatic disease. However, photosensitivity may have a significant impact on quality of life of EPP patients.
- 24. TÍTULO: Erythropoieticprotoporphyria AUTORES: Mario Lecha, Hervé Puy, and Jean Charles Deybach LINK: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2747912/?tool=pubmed
- 27. 4. ABSTRACT Although porphyria is a relatively uncommon condition, it should be considered in patients presenting with an atypical medical, psychiatric, or surgical history. Acute attacks are associated with a substantial morbidity and mortality; there is a need for rapid and accurate diagnosis of the neuropsychiatric porphyrias, particularly because haemarginate can induce a definite remission if given early in an attack. Additionally, porphyrias may present with skin lesions or photosensitivity.
- 28. TITULO: Diagnosis and management of porphyria AUTORES: Helen Thadani, specialistregistrar,a Allan Deacon, consultantclinicalscientist,b and Timothy Peters, professor LINK: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1127427/?tool=pubmed
- 31. 5. ABSTRACT Porphyriasare metabolic diseases resulting from deficiencies of specific enzymes of the heme biosynthetic pathway. These enzymes are most active in bone marrow and liver. When these diseases are manifest, accumulation of 1 or more intermediates occurs initially in 1 of these tissues.
- 32. Erythropoieticporphyrias, in which overproduction of heme pathway intermediates occurs primarily in bone marrow erythroid cells, usually present at birth or in early childhood with cutaneous photosensitivity, or in the case of congenital erythropoieticporphyria, even in utero as nonimmunehydrops.
- 33. Most porphyrias are hepatic, with overproduction and initial accumulation of porphyrin precursors or porphyrins occurring 1st in the liver. Regulatory mechanisms for heme biosynthesis in liver are distinct from those in the bone marrow and appear to account for activation of hepatic porphyrias during adult life rather than childhood.
- 34. Homozygous forms of the hepatic porphyrias may manifest clinically prior to puberty, and asymptomatic heterozygous children may present with nonspecific and unrelated symptoms. Parents often request advice about long-term prognosis and information about management of these disorders and drugs that can be taken safely to treat other common conditions.
- 35. TITULO: Porphyria AUTORES: Karl E. Anderson, Chul Lee, Robert J. Desnick LINK: http://biblioteca.fucsalud.edu.co:2052/books/page.do?sid=1120246258&eid=4-u1.0-B978-1-4160-2450-7..50093-1&isbn=978-1-4160-2450-7&type=bookPage§ionEid=4-u1.0-B978-1-4160-2450-7..50093-1--cesec4&uniqId=235811794-9
- 37. LIBRO
- 38. Las porfirias son trastornos metabólicos y cada una de ellas es consecuencia de la deficiencia de alguna enzima específica en la vía de biosíntesis del grupo hemo. Las deficiencias enzimáticas se heredan por medio de rasgos autosómicos dominantes o recesivos, con excepción de la porfiria cutánea tardía (PCT), que suele ser esporádica.
- 39. Las porfirias se clasifican en hepáticas o eritropoyéticas, según el sitio primario de producción excesiva y acumulación de sus precursores respectivos porfirínicos o porfirinas, aunque algunas comparten características comunes. Por ejemplo, la PCT que es la porfiria más frecuente, es de tipo hepático y su sintomatología inicial incluye fotosensibilidad cutánea ampollosa.
- 40. Las principales manifestaciones de las porfirias de tipo hepático agudas son neurológicas y comprenden dolor abdominal neuropático, neuropatía motora periférica y perturbaciones psíquicas y sus ataques suelen ser desencadenados por hormonas esteroides, algunos fármacos e influencias nutricionales.
- 41. Las porfirias de origen hepático generan síntomas principalmente en los adultos, pero se han identificado variantes homocigotas raras de las porfirias autonómicas dominantes de tipo hepático que por lo general surgen clínicamente antes de la pubertad.
- 42. TITULO Porfirias Autores Robert J. Desnick, Kenneth H. Astrin Capítulo 352. Porfirias