2. Capacidad calorífica , C
Es una propiedad extensiva que indica la cantidad de calor
necesaria para elevar la temperatura de un cuerpo,1ºC o 1K.
Depende de su masa y composición. Unidades: J/ºc, cal/ºC
Capacidad calorífica molar ( )
Es la capacidad calorífica de un mol de sustancia.
Unidades: J/mol. ºC, cal/mol.ºC
Capacidad calorífica específica (ce)
Es la capacidad calorífica de un gramo de sustancia
(calor específico). Unidades: J/g. ºC, cal/g.ºC
C
OH2 1 / .º 18 / .º 4,184 / .ºce cal g C cal mol C J g C
4. TIPOS DE CAPACIDAD CALORÍFICA
1. Capacidad calorífica a presión constante (Cp)
dT
dH
dT
dq
C p
p dcTbTaTCp 23
2
1
T
T
pp TdCqH
TCnq pp
Tcemqp
TCqp
5. Calorímetro a presión constante
Se puede utilizar para determinar los calores de
reacción diferentes de la combustión.
0 solcalrxnsistema qqqq
)( calsolrxn qqq
Para soluciones diluidas:
ce (sol) = ce (agua) = 1 cal/g.ºC
Procesos:
• Neutralización
• Calores de solución
• Calores de dilución
)( TCTcemq calsolsolrxn
6. TIPOS DE CAPACIDAD CALORÍFICA
2. Capacidad calorífica a volumen constante (Cv)
dT
dU
dT
dq
C v
v dcTbTaTCv 23
2
1
T
T
vv TdCqU
TCnq vv
Tcemqv
dTCnq
T
T
vv
2
1
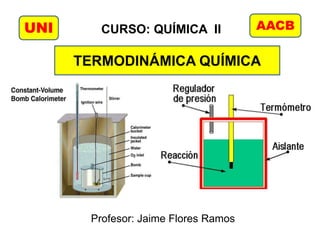
7. Calorímetro a volumen constante (bomba calorimétrica)
Se puede utilizar para determinar los calores de
reacción de combustión.
0 aguacalrxnsistema qqqq
)( calaguarxn qqq
)( TCTcemq calaguaaguav La combustión de 1 g de ácido benzoico
produce 26,38 kJ de calor con el cual se
puede calcular la capacidad calorífica
del calorímetro
8. Relación entre Cp y Cv
H = U + PV (definición)
d PVdH dU
dT dT dT
dT
PVd
CC vp
PV = n R T RTVP
R
dT
RTd
dT
VPd
RCC vp (para n = 1 mol)
a. Para gases ideales:
9. b. Para sólidos o líquidos:
KmolJRCv ./471,12
2
3
)(aproxCCpequeño
dT
VPd
vp
c. Para gases monoatómicos
KmolJRC p ./78,20
2
5
d. Para gases diatómicos
KmolJRCv ./786,20
2
5
KmolJRC p ./099,29
2
7
10. ENTALPÍA ESTÁNDAR DE LA REACCIÓN, ΔHº
ΔHº = npHf
0
(productos)– nrHf
0
(reactivos)
• Es el calor liberado o absorbido por el sistema durante el
proceso a condiciones estándar.
• Es la diferencia entre la entalpía de los productos y la
entalpía de los reactivos.
a A + b B → c C + d D ΔHº
)( ººººº
BADCrxn HbHaHdHcH
11. Dependencia de ΔH con respecto a la temperatura
a A + b B → c C + d D
dTCHH
T
T
p
2
1
º
1
º
2
ΔH cambia muy poco con el cambio de temperatura
ΔCp = Cp(productos) - Cp (reactivos)
ΔCp = c Cp(C) + d Cp (D) - (a Cp (A) + b Cp (B))
12. ΔH1
ΔH”ΔH´
ΔH2
a A + b B
a A + b B c C + d D
c C + d D
(T2)
(T1)
ΔH1 = ΔH´ + ΔH2 + ΔH”
dTCHdTCH
T
T
productosp
T
T
reactivosp
1
2
2
1
)(2)(1
dTCHH
T
T
p
2
1
º
1
º
2
13. Temperatura teórica de
llama o flama
•Es la máxima temperatura que se puede
obtener cuando se quema el gas con la
cantidad de aire establecido, considerando
un sistema adiabático.
• Con el fin de calcular la temperatura de
llama se empleará cualquier trayectoria entre
los estados inicial y final.
14. Gráfica de la temperatura contra la energía térmica añadida
cuando 1 g inicialmente a –30°C se convierte en vapor a
120°C.
Hielo
Hielo + agua
Agua
Agua +
vapor
Vapor
62.7 396.7 815.7 3076
-30
0
50
100
T(°C)
A
B
C
D
E
Se
calienta
el hielo
Se funde
el hielo
Se
calienta
el agua
Se
evapora
el agua
Se
calienta
el vapor
120
15. PROCESOS TERMODINÁMICOS
1. PROCESOS ISOBÁRICOS
P = cte
2. PROCESOS ISÓCOROS
V = cte
3. PROCESOS ISOTÉRMICOS,
T = cte
4. PROCESOS ADIABÁTICOS,
Q = 0
(No entra ni sale calor del sistema)
16. PROCESOS TERMODINÁMICOS
1. Proceso isobárico
P = cte.
1 2
V1 V2
P
W = - P ∆V = - P (V2 - V1)
∆H = Qp = n Cp ∆T
V1 = V2
T1 T2
∆U = Qp + WV2V1
P
17. 2. Proceso isocórico, isométrico V = cte
W = 0
∆U = Qv = n Cv ∆T
P1 = P2
T1 T2
p
V
P1
P2
18. 3. Proceso isotérmico, T = cte
1
2
V1
P1
P2
W = - ∫ PdV = - nRT Ln(V2/V1)
= -2,3 nRT ℓog(V2/V1)
P1 V1 = P2V2
W = - nRT Ln(P1/P2)
∆U= 0 ∆H = 0
Q = - W
p
VV1
V2
20. 4. Proceso adiabático, Q = 0
∆E = W = n Cv ∆T
En estos casos el trabajo realizado será a costa de la energía interna
W = n Cv(T2 - T1)
W = n Cv(P2V2/nR - P1V1/nR)
W = Cv/R(P2V2 - P1V1)
Cp = Cv + R
W = Cv (P2V2 - P1V1)
Cp-Cv
W = ∆PV = n R ∆T
γ – 1 γ – 1
21.
22. Proceso espontáneo, exergónico
• Significa capaz de suceder, sin necesidad de trabajo
para lograrlo.
• No significa rápido.
• En cualquier proceso espontáneo, el camino entre
reactivos y productos es irreversible.
26. Proceso reversible
• Es posible devolver al sistema y su entorno al estado inicial
por el mismo camino.
• Es una sucesión de estados de equilibrio del sistema con su
entorno.
• Las funciones termodinámicas cambian muy lentamente de
un momento a otro.
• La dirección del proceso se puede invertir en cualquier
momento, haciendo un cambio infinitesimal (muy pequeño)
en el medio ambiente.
Ideal
Proceso irreversible Real
• Las funciones termodinámicas cambian considerablemente
de un momento a otro.
• No pueden ser detenidos ni invertidos por un cambio
infinitesimal de las condiciones externas.
27. Los procesos termodinámicos realizados en
la naturaleza son irreversibles, es decir hay
una dirección en la que el proceso se realiza
espontáneamente, pero lo contrario no es
espontaneo
El ingeniero se esfuerza constantemente en
reducir la irreversibilidad de los sistemas
para obtener un funcionamiento mejor de
estos.
28. ENTROPÍA (S)
• Indica el grado de
desorden molecular.
• Es una propiedad
extensiva, función de
estado, que mide la
dispersión caótica de
la energía.
• Depende de la
temperatura, presión y
composición.
Entropía creciente
29. CAMBIO DE ENTROPÍA, ∆S
• Es el calor transferido al sistema durante un trayecto
reversible del estado 1 al estado 2.
∆S = ∫ dqrev = qrev
T T
q rev = Calor agregado al sistema, mientras éste pasa de 1 a 2
Segunda ley de la termodinámica
a) En un proceso reversible, no espontáneos (Endergónicos)
S universo = cte ∆S universo = 0
b) En un proceso irreversible, espontáneos ( Exergónicos)
S universo > 0 ∆S universo > 0
31. 1. Para cambio de estado físico
a) Fusión, proceso reversible a T = cte
∆S = qrev = ∆Hfusión
T T fusión
H2O (s) H2O (ℓ) ∆ H = 6,02 kJ/mol y Tf = 0 ºC
∆S = 22 J/K
b) Ebullición, proceso reversible a T = cte
∆S = qrev = ∆Hebullición
T T ebullición
H2O (ℓ) H2O (g) ∆ H = 40,67 kJ/mol y Te = 100 ºC
∆S = 109 J/K
32. 2. Para compresión o expansión isotérmica
de un gas ideal
• La energía interna de un gas ideal depende
únicamente de la temperatura.
Ec = 3/2 RT
∆S = q rev
T
Pero: ∆U= Q + W Q = - W
∆S = nRLn(V2/V1) ∆S = nRLn(P1/P2)
Expansión (V2 > V1) ∆S > 0
Si
Compresión (V2<V1) ∆S < 0
33. 3. Dependencia de la entropía y la
temperatura
Pero: dqrev = nCp dT o dqrev = nCv dT
∆S = nCpLn(T2/T1) ∆S = nCvLn(T2/T1)
Si
T2 > T1 ∆S > 0
T2 < T1 ∆S < 0
34. Tercera ley de La termodinámica
• La entropía de los cristales perfectos de los
elementos o compuestos puros es cero a la
temperatura del cero absoluto, 0 K.
S = 0 ( Orden perfecto)
35. Cambio de entropía para una reacción
Química, ∆S
a A + b B c C + d D
∆S = c Sº (C) + d Sº (D) - ( a Sº (A ) + b Sº ( B))
Hallar el cambio de entropía estándar a 25 ºC , para la reacción
siguiente:
4 Fe (s) + 3 O2 (g) 2 Fe2O3 (s) ∆Hº = - 1648,4 kJ
∆Sº = 2 mol(87,4 J/mol.K) - 4 mol(27,3 J/mol.K) +
3 mol(205 J/mol.K)
∆Sº = - 549,4 J/ K
36. Cambio de entropía del entorno, ∆S
∆Sent = - ∆Hsistema
T
Exotérmico (∆H < 0) ∆S ent > 0
Si la reacción
Endotérmico (∆H > 0) ∆Sent < 0
En la reacción:
∆S ent = - ∆H/T = - (- 1648,4 kJ/298 K)
∆S ent = 5531 J/K
∆S univ = - 549,4 J/ K + 5531 J/K = 4982 J/K
Como ∆S univ > 0 La oxidación del hierro, es espontáneo
37. ENERGÍA LIBRE DE GIBBS, ∆G
• Se llama también función de Gibbs.
• Es una función de estado, propiedad extensiva.
• Es aquella energía útil del sistema a presión constante que
puede transformarse en trabajo.
• Nos proporciona información valiosa acerca de la
espontaneidad de procesos a T y P constantes.
Se sabe que un proceso es espontáneo, cuando:
∆S > 0 La energía libre de Gibbs resume.
∆H < 0
∆G = ∆H - T ∆S
T = Temperatura absoluta en, K
38. Josiah Willard Gibbs
(1839-i1903) Físico y químico estadounidense. A la edad de quince años ingresó en la
Universidad de Yale, donde obtuvo el primer doctorado en ingeniería concedido por la
mencionada institución.
Durante un viaje a Europa, entró en contacto con los físicos y matemáticos de mayor
prestigio de la época, cuyas novedosas aportaciones estudió con interés. Centró durante un
tiempo su atención en el estudio de la máquina de vapor de Watt; ocupado en el análisis del
equilibrio de la máquina, Gibbs empezó a desarrollar un metódo mediante el cual podia
calcular las variables involucradas en los procesos de equilibrio químico.
Dedujo la regla de las fases, que permite determinar los grados de libertad de un sistema
fisicoquímico en función del número de componentes del sistema y del número de fases en
que se presenta la materia involucrada.
También definió una nueva función de estado del sistema termodinámico, la denominada
energía libre o energía de Gibbs (G), que permite prever la espontaneidad de un
determinado proceso fisicoquímico (como puedan ser una reacción química o bien un
cambio de estado) experimentado por un sistema sin necesidad de interferir en el medio
ambiente que le rodea.
En 1871 fue designado profesor de física matemática en Yale, tras la publicación de su labor
fundamental, que incluyó los títulos Métodos gráficos en termodinámica de fluidos y Sobre
el equilibrio de sustancias heterogéneas, este último de importancia trascendental para la
posterior evolución de la física y la química moderna.
La descripción adecuada de los procesos termodinámicos desde el punto de vista de la
física llevó a Gibbs a desarrollar una innovadora herramienta científica, la mecánica
estadística, que con posterioridad se reveló útil para la moderna mecánica cuántica.
39. ∆G < 0 La reacción es espontánea en el
sentido establecido.
∆G > 0 La reacción no es espontánea en el
sentido establecido.
∆G = 0 El sistema está en equilibrio.
40. CAMBIO DE ENERGÍA LIBRE ESTÁNDAR, ∆Gºf
• La energía libre estándar de formación de
Gibbs, son los valores cuando se forma una
cantidad unitaria de sustancia a partir de sus
elementos en estado estándar a la temperatura
especificada, generalmente 25 ºC
• La energía libre de formación de los elementos
en su estado estándar es cero.
La energía libre estándar de formación de
Gibbs a 25 ºC, ∆Gºf en kJ/mol
SÓLIDO LÍQUIDO GAS
NaCl - 384,0 H2O - 237,2 NH3 - 16,5
KCl - 408,3 CH3OH - 166,4 CO2 - 394,5
41. PARA UNA REACCIÓN QUÍMICA
∆G0
reacción = ∑n ∆G0
productos – ∑n ∆G0
reactivos
Calcular la variación de la energía libre a 25 0C y 1 atmósfera
de presión para la siguiente reacción y establecer si es o no
espontánea.
CH4(g) + 2O2(g) → CO2(g) + 2H2O(l)
∆G0 = -32,89 kJ/mol ∆G0 = 0 ∆G0 = -394,4 kJ/mol ∆G0 = -237,2 kJ/mol
∆G0
rex = 1mol(-394,4 kJ/mol) +2 mol(-237,2 kJ/mol) – 1mol(-
32,89kJ/
∆G0
rex = -868.8 kJ – (-32.89 kJ)
∆G0
rex = -835.91 kJ
∆G0
reacción < 0 Reacción espontánea
42. Si la reacción se lleva a cabo a otra temperatura, es
necesario hacer una corrección y se utiliza:
∆G0
reacción = ∆H0
reacción – T∆S0
reacción
Si la reacción se realiza a 400 K, se calcula:
∆H0
reacción = -890.4 kJ
∆S0
reacción = 353.4 J/K – 186.19 J/K =167.21 J/K
Nota: El valor de S0 por ser pequeño está reportado en J/K mol
∆G0
reacción = ∆H0
reacción – T∆S0
reacción
∆G0
reacción = -957.28 Reacción espontánea
43. Ejercicios
Calcular la variación de la energía libre de Gibbs
para las reacciones siguientes a 25 0C y 200 0C y
decir si son o no espontáneas a estas temperaturas:
a) CO2(g) + H2O(l) → C6H12O6(s) + O2(g)
b) C2H2(g) + O2(g) → CO2(g) + H2O(l)
c) 2H2O(l) → 2 H2(g) + O2(g)
44. Espontaneidad de un proceso
∆H ∆S ∆G
+ + Espontáneo, sólo a elevada temperatura
- + Espontáneo a cualquier temperatura
+ - No espontáneo a cualquier temperatura
- - Espontáneo, sólo a baja temperatura
∆G = ∆H - T∆S