Download to read offline






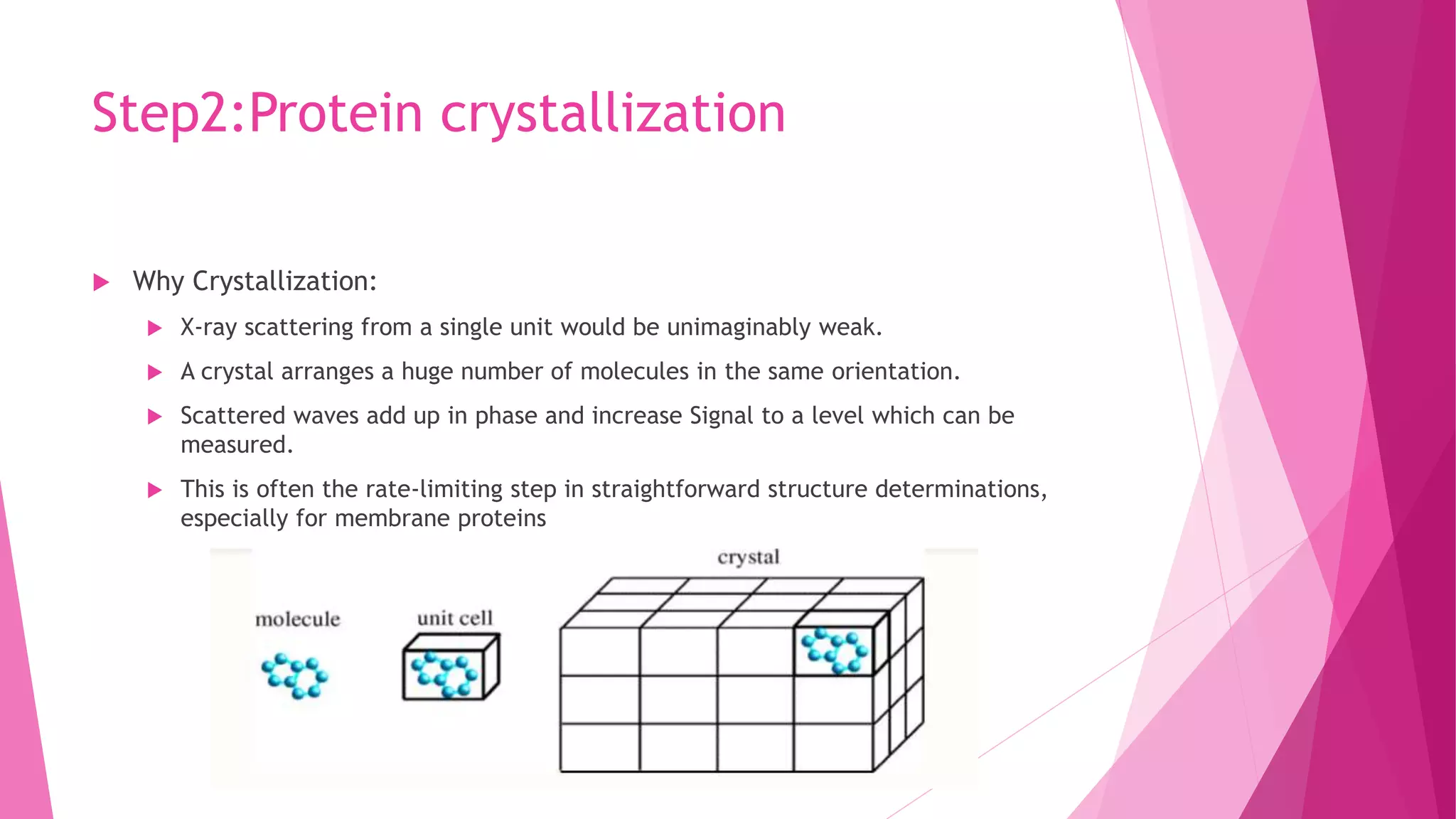
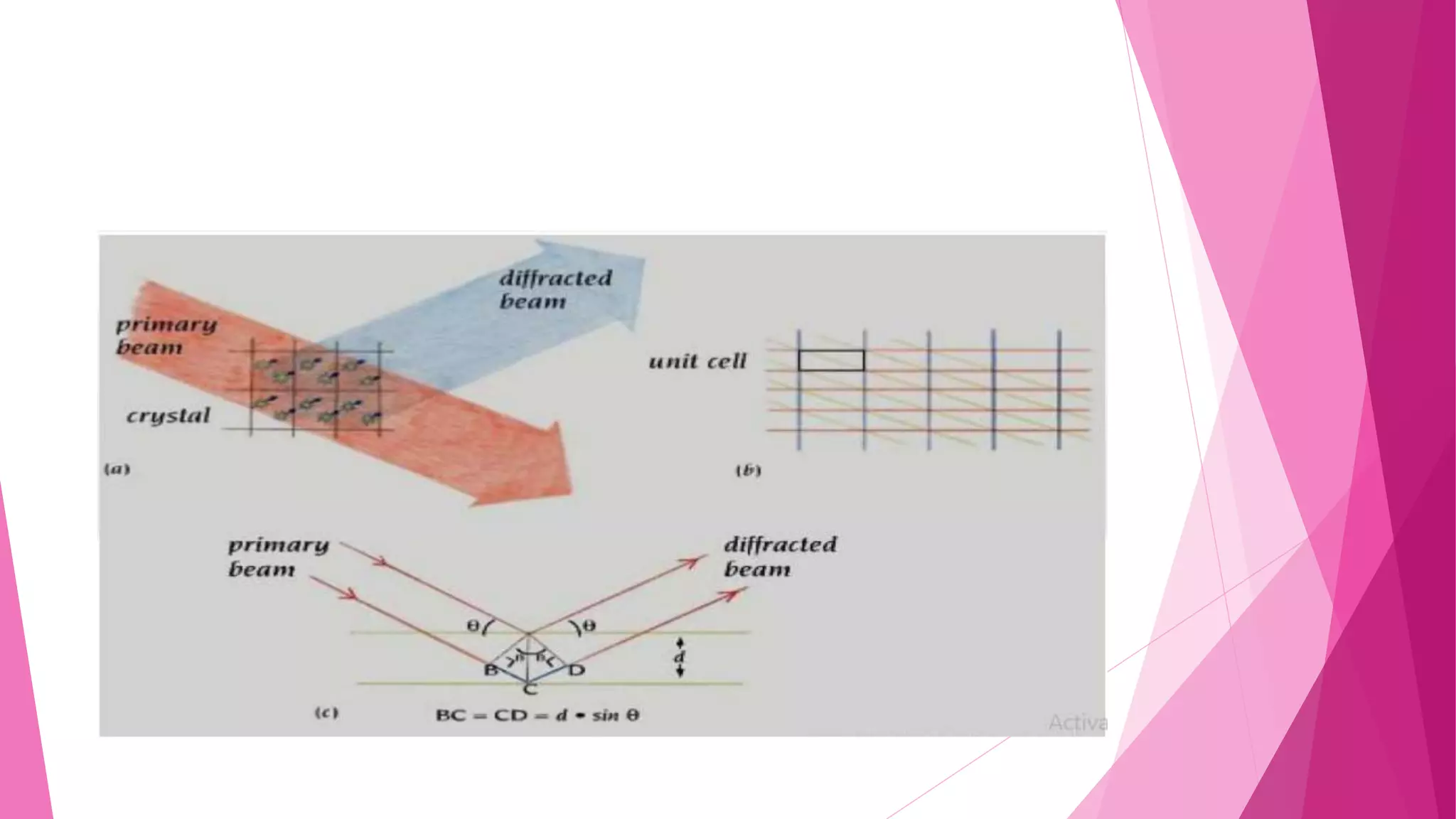

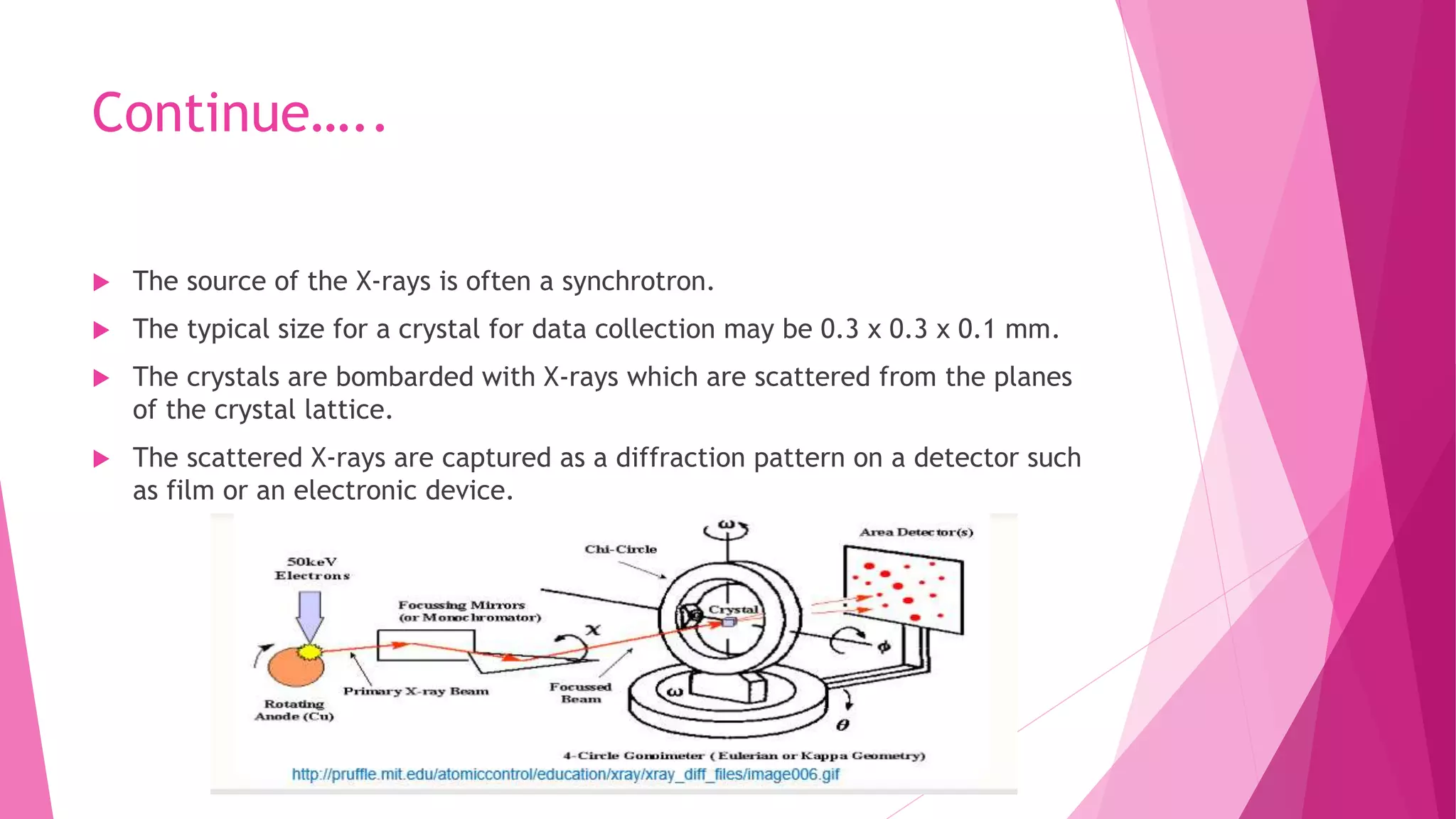












This document provides an overview of X-ray crystallography, a technique used for visualizing protein structures at the atomic level through the diffraction of X-rays by crystalline materials. The process involves several critical steps including protein purification, crystallization, data collection, and structure determination, with various methods to resolve the phase problem. The applications of X-ray crystallography are highlighted, including its role in studying HIV protease and developing effective painkillers for arthritis.



































































