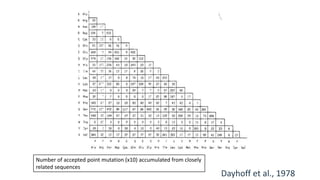
The document discusses the limitations of identity scoring in protein sequence alignment, particularly highlighting its poor diagnostic power due to equal weighting of matches and reliance on simplistic scoring. It introduces the Point Accepted Mutation (PAM) model, conceptualized by Margarett Dayhoff, which provides a more reliable framework for understanding evolutionary changes in proteins through a mutation probability matrix. Additionally, the document details how PAM matrices are constructed, the significance of various amino acid substitutions, and the application of odds matrices in evaluating sequence relationships.

















































![Bio info statistical-methods[1]](https://cdn.slidesharecdn.com/ss_thumbnails/bioinfo-statisticalmethods1-150123025112-conversion-gate02-thumbnail.jpg?width=640&height=640&fit=bounds)






















![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)










