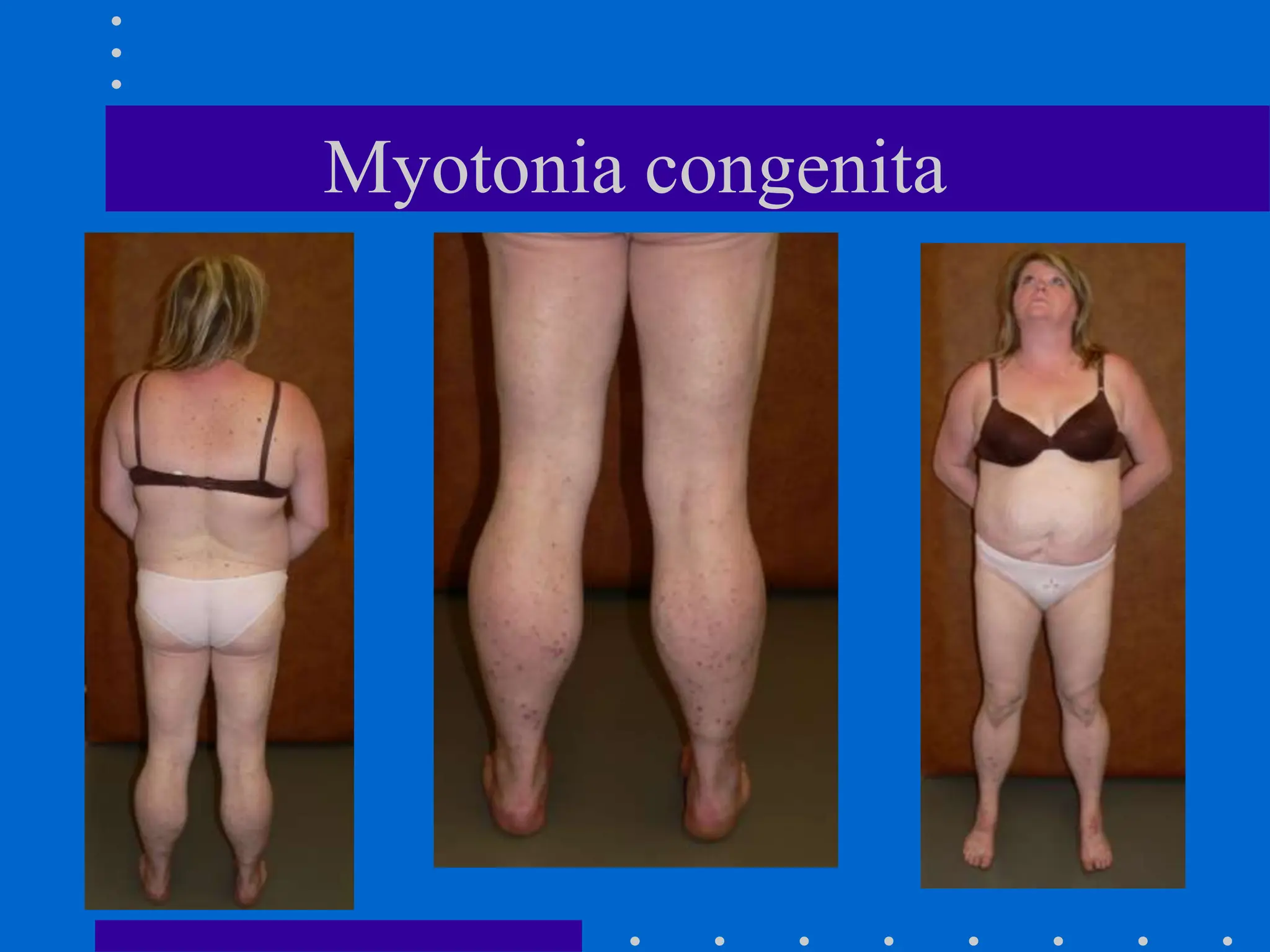
The document provides a comprehensive overview of neuromuscular disorders, focusing on various types of myopathies and their symptoms, diagnoses, and treatments. Key conditions discussed include Duchenne muscular dystrophy, myotonic dystrophy, inflammatory myopathies, and myasthenia gravis, along with their clinical features and management strategies. The document emphasizes the importance of genetic and biochemical testing in diagnosing these disorders and outlines potential therapeutic approaches.