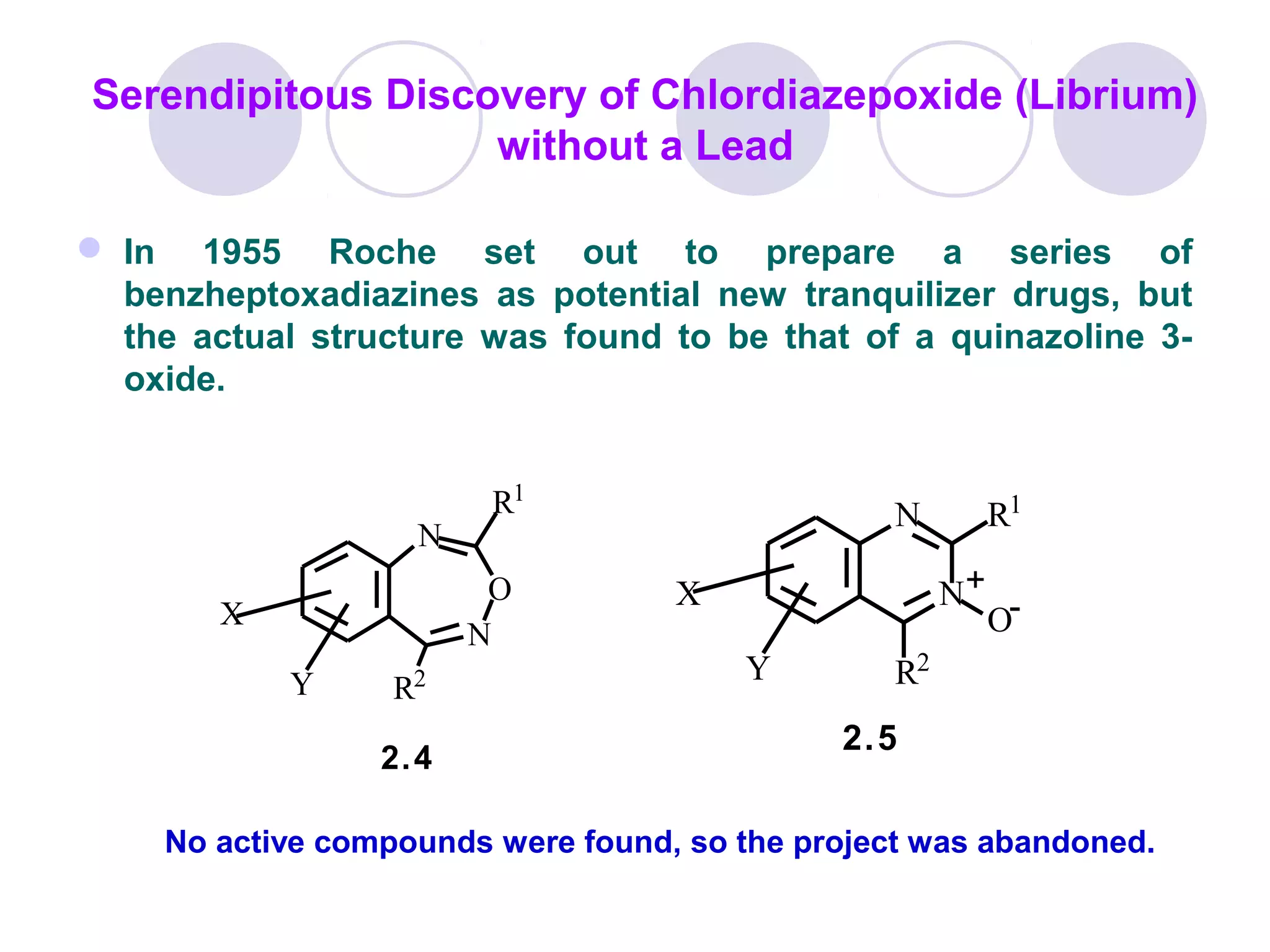
Drug design involves finding new medications by understanding how they interact with biological targets at a molecular level. The goal is to design small organic molecules that either activate or inhibit biomolecules like proteins. Effective drug design optimizes properties like binding affinity as well as bioavailability, toxicity, and side effects. The process involves identifying lead compounds, determining their structures and effects on biological activity, and using modeling to design analogs with improved target interactions and safety profiles. Overall, drug design and development is a lengthy process taking over 10 years and costing hundreds of millions to bring a new medication to market.