Downloaded 73 times





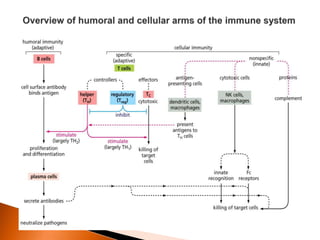





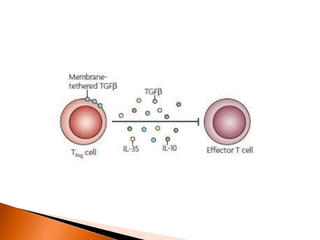

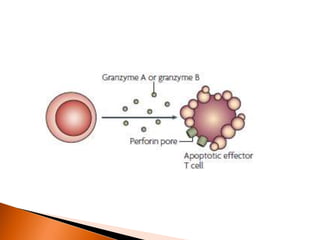


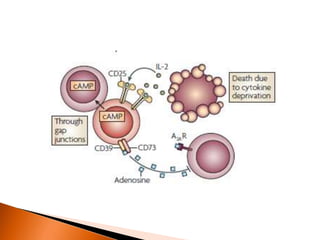


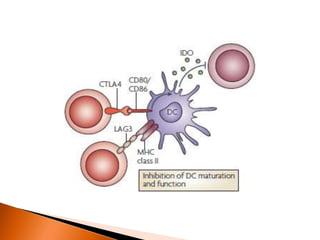






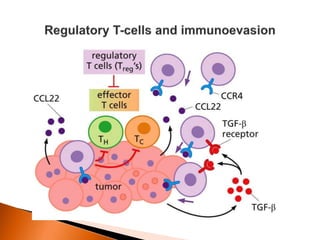












Regulatory T-cells (Tregs) help maintain self-tolerance and prevent autoimmunity by suppressing immune responses. They express FOXP3 and CD25 and function through various mechanisms like secreting inhibitory cytokines or metabolizing IL-2. Tregs are implicated in tumor immune escape by suppressing anti-tumor immunity. While Tregs are normally beneficial, in cancer high levels associate with poor prognosis by hindering immune response. Emerging immunotherapies aim to deplete or modulate Tregs to enhance anti-tumor immunity.
















































![Chapter 39 role of radiotherapy in benign diseases.pptx [read only]](https://cdn.slidesharecdn.com/ss_thumbnails/chapter39roleofradiotherapyinbenigndiseases-191105205437-thumbnail.jpg?width=640&height=640&fit=bounds)






















![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)






![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)








