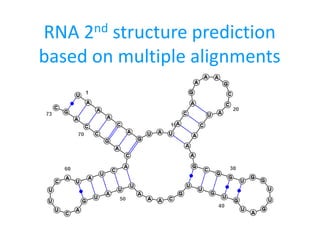
2. RNA evolution
● Homologous RNAs can have a common 2nd structure
without sharing a significant sequence similarity
● Mutations can lead to compensatory mutations to maintain
the base-paring complementarity
3. Comparative sequence analysis
● In a structurally correct multiple alignment of RNAs,
conserved base pairs are often revealed by the presence of
frequent correlated compensatory mutations
● Measure sequence covariation: mutual information
● is the frequency of one of the four bases observed in col I
● is the joint frequency of the base pairs observed in
columns i and j
𝑀𝑖𝑗 = ∑
𝑥 𝑖,𝑥 𝑗
𝑓𝑥 𝑖,𝑥 𝑗
log2
𝑓𝑥 𝑖,𝑥 𝑗
𝑓𝑥 𝑖
⋅ 𝑓𝑥 𝑗
𝑓𝑥 𝑖
𝑓𝑥 𝑖,𝑥 𝑗
4. Covariance method
G U C U U C G G A C
G A C U U C G G U C
G G C U U C G G C C 𝑀2,9 = 3 ⋅
1
3
⋅ log2
1/3
1/9
= log23 ≈ 1.59
𝑀𝑖𝑗 = ∑
𝑥 𝑖,𝑥 𝑗
𝑓𝑥 𝑖,𝑥 𝑗
log2
𝑓𝑥 𝑖,𝑥 𝑗
𝑓𝑥 𝑖
⋅ 𝑓𝑥 𝑗
● Mij varies between 0 and 2
● Mij is 2 when i and j appear completely random but are perfectly
correlated
● if i and j are uncorrelated, the mutual information is 0
● if either i or j are highly conserved positions, we also get little or
no mutual information
5. ● Mij is 2 when i and j appear completely random but are perfectly
correlated
● if i and j are uncorrelated, the mutual information is 0
● if either i or j are highly conserved positions, we also get little or
no mutual information
Covariance method
G U C U U C G G A C
G A C U U C G G U C
G G C U U C G G C C
G C C U U C G G G C
𝑀1,9 = 4 ⋅
1
4
⋅ log2
1/4
1/4
= 0
𝑀𝑖𝑗 = ∑
𝑥 𝑖,𝑥 𝑗
𝑓𝑥 𝑖,𝑥 𝑗
log2
𝑓𝑥 𝑖,𝑥 𝑗
𝑓𝑥 𝑖
⋅ 𝑓𝑥 𝑗
𝑀2,9 = 4 ⋅
1
4
⋅ log2
1/4
1/16
= 2
6. Comparative analysis
● Start with a multiple alignment
● Predict 2nd structure base on alignment
● Refine alignment based on 2nd structure
● Repeat
● The sequences to be compared must be sufficiently:
● similar that they can be initially aligned by primary sequence
● dissimilar that a number of covarying substitutions can be
detected
7. Comparative analysis
● How to build 2nd structure based on alignment?
● Greedy method
● choose the pair of columns that have the highest Mij
● make a base pairs
● carry on with the second highest Mij
● problem columns might end up in more than one base pair
8. SCFGs and RNA alignments
● An SCFG could be modified to generate columns of
alignments instead of nucleotides
● Requires a fixed number of sequences in the alignment
● Instead, change it to generate the structure!
𝑆 → . 𝑆 ∣ 𝑆.
𝑆
𝑆𝑆
ε
𝑆 → 𝑎𝑆 ∣ 𝑐𝑆 ∣ 𝑔𝑆 ∣ 𝑢𝑆
𝑆𝑎 ∣ 𝑆𝑐 ∣ 𝑆𝑔 ∣ 𝑆𝑢
𝑎𝑆𝑢 ∣ 𝑐𝑆𝑔 ∣ 𝑔𝑆𝑢
𝑢𝑆𝑎 ∣ 𝑔𝑆𝑐 ∣ 𝑢𝑆𝑔
𝑆𝑆
ε
9. SCFGs and RNA alignments
● How to determine the probability of a structure for a given
sequence?
● A C G U C G U C
● ( ( ( . ) ) ) .
● Use CYK to calculate the maximum probability of a
structure for a given sequence...
𝑆 ⇒ 𝑆. ⇒ 𝑆 . ⇒ 𝑆 . ⇒ 𝑆 . ⇒ . 𝑆 . ⇒ . .
10. SCFGs and RNA alignments
● Use a phylogenetic tree (including branch lengths) to:
● determine the probability of a column to be single
● determine the probability of two columns to form a base pair
● Use the SCFG and the columns probability to determine the
best secondary structure for the alignment
● CYK and the other SCFGs algorithms are basically the same