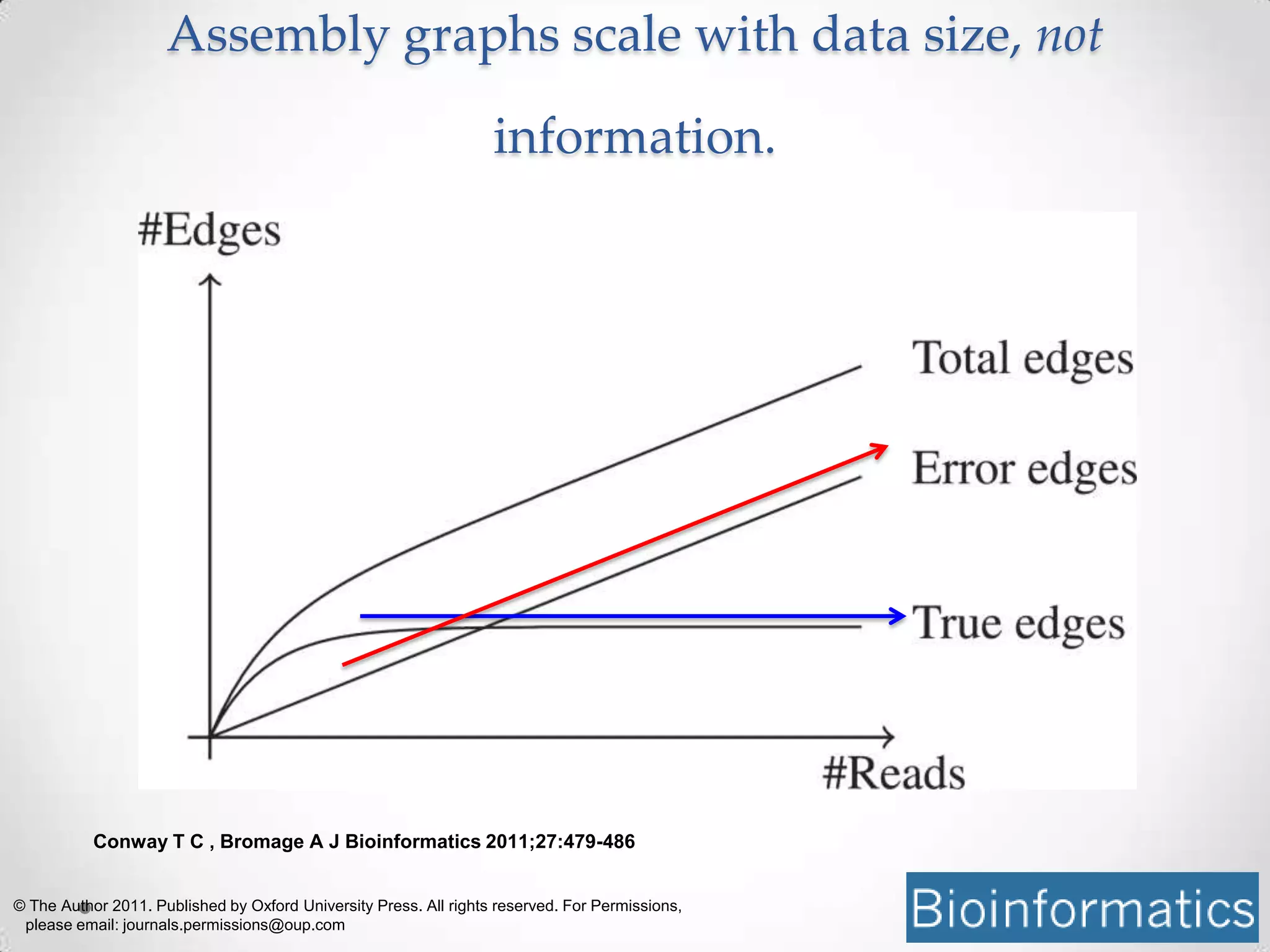
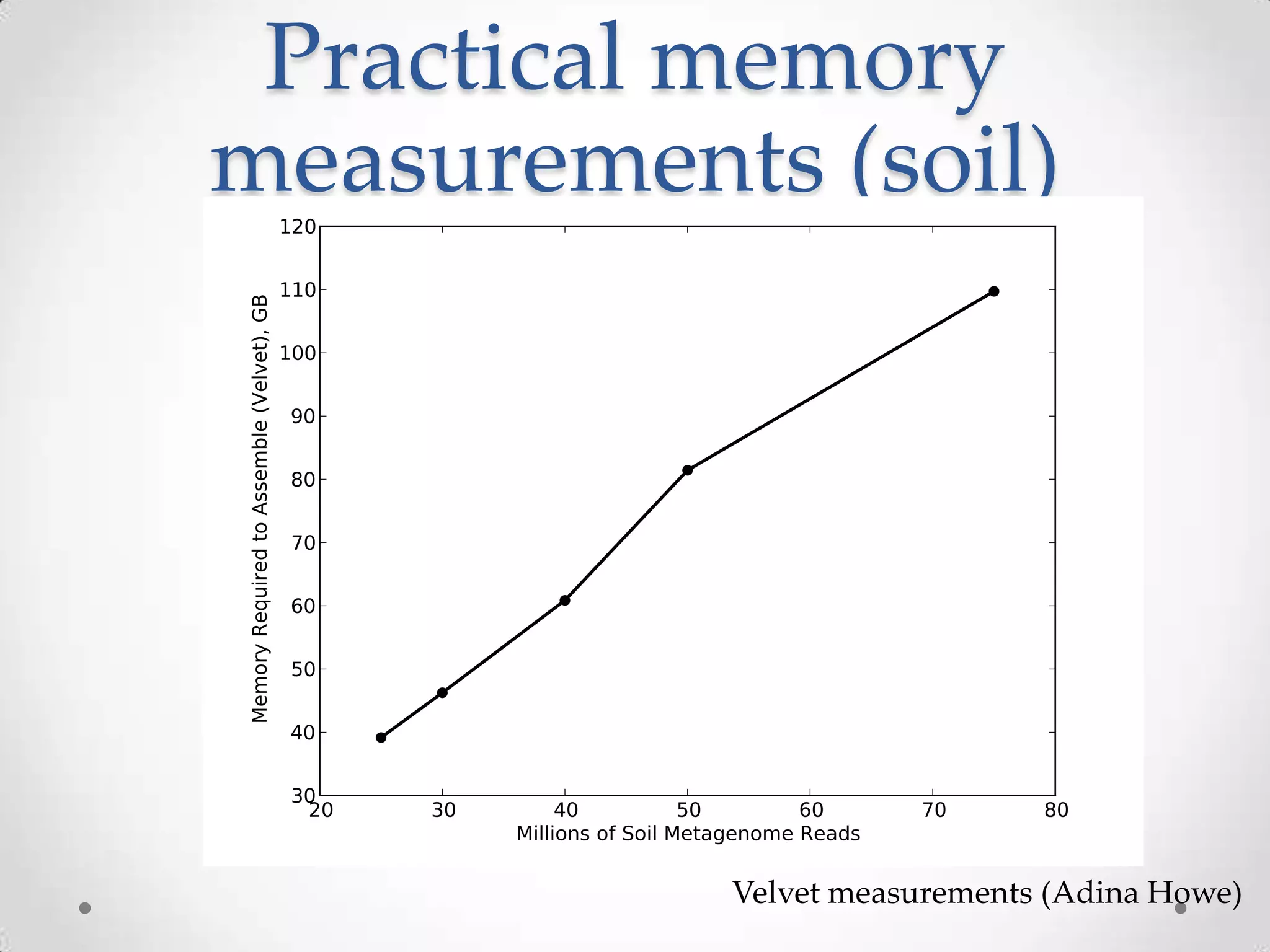
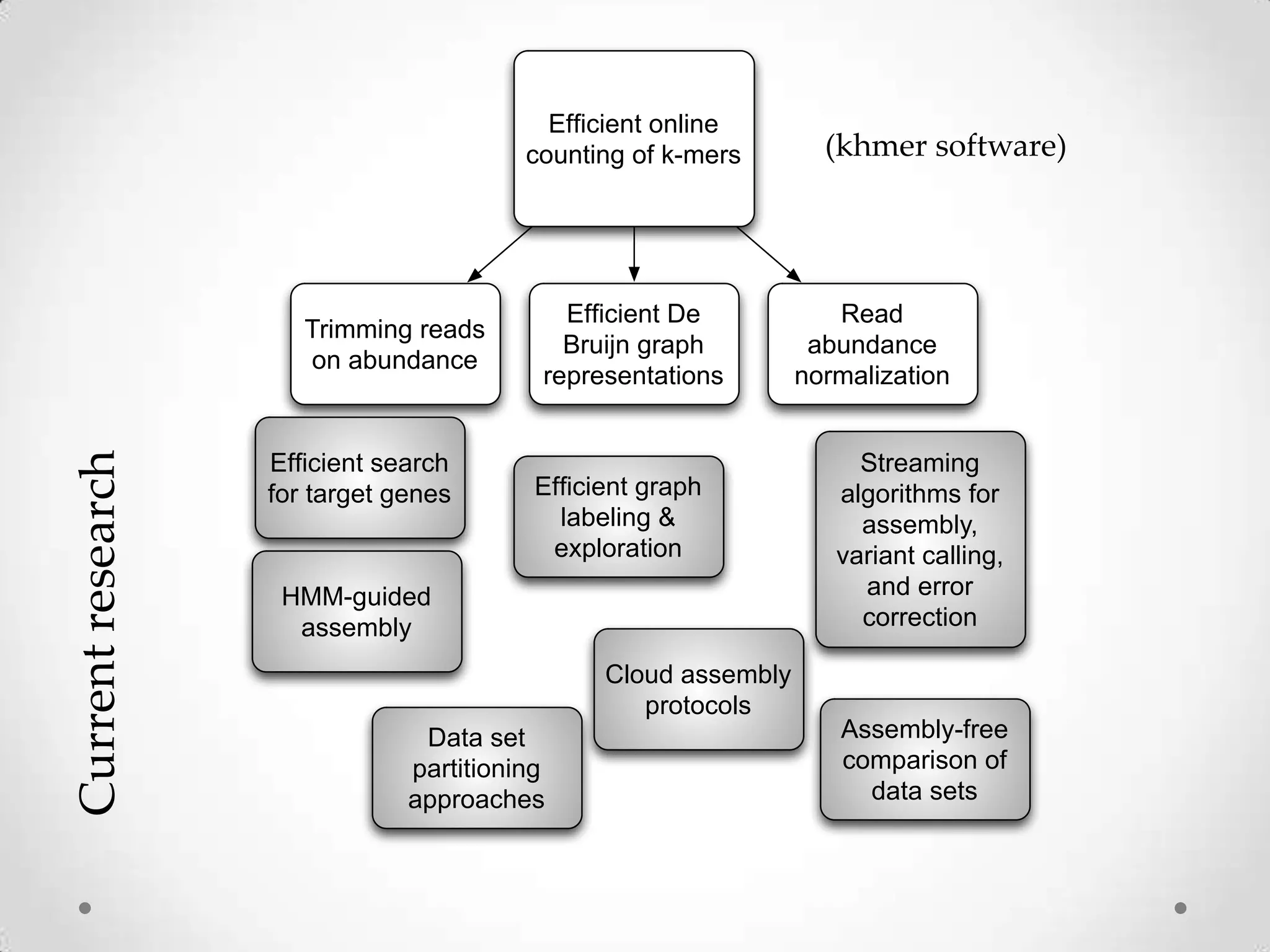
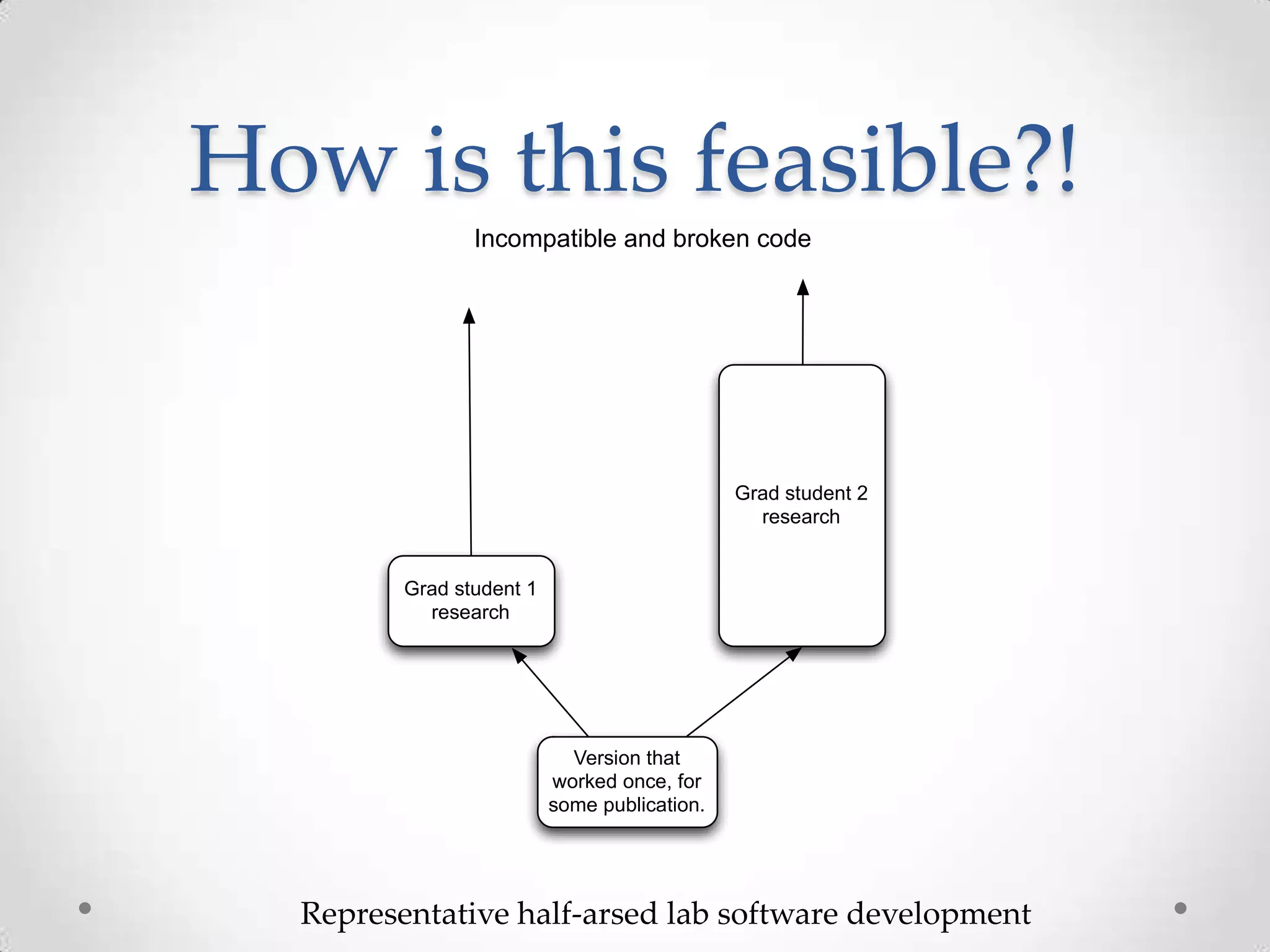
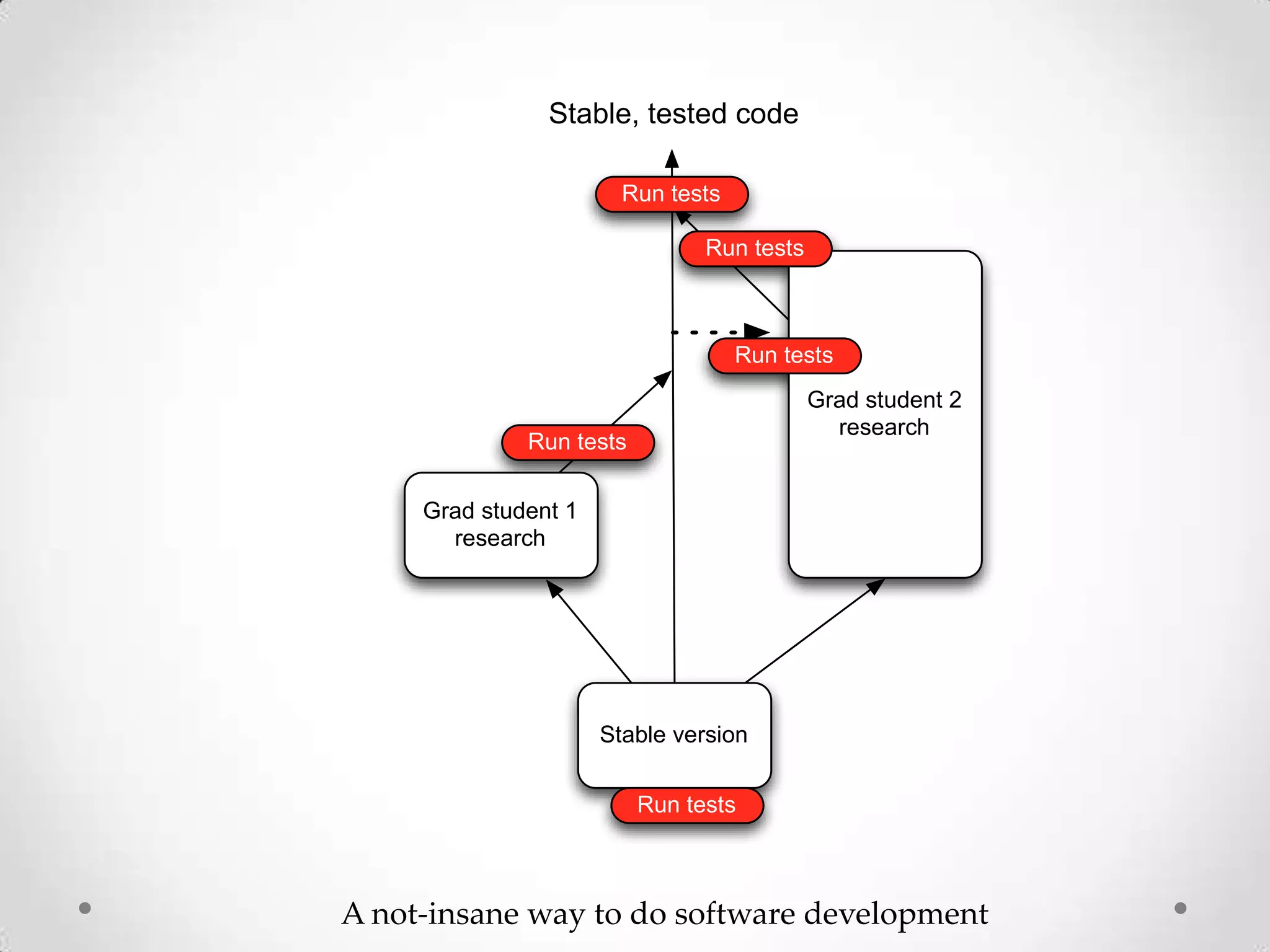
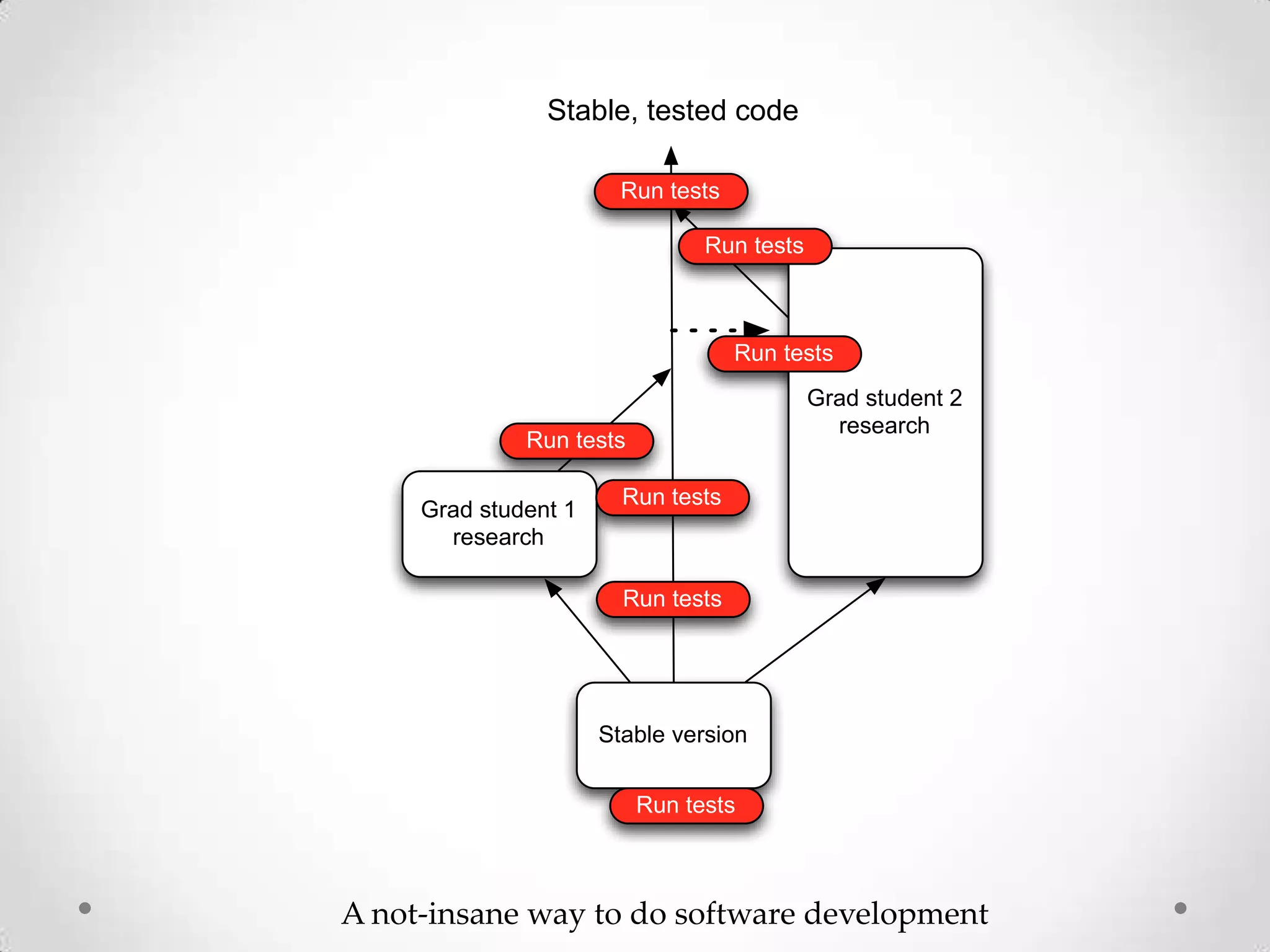
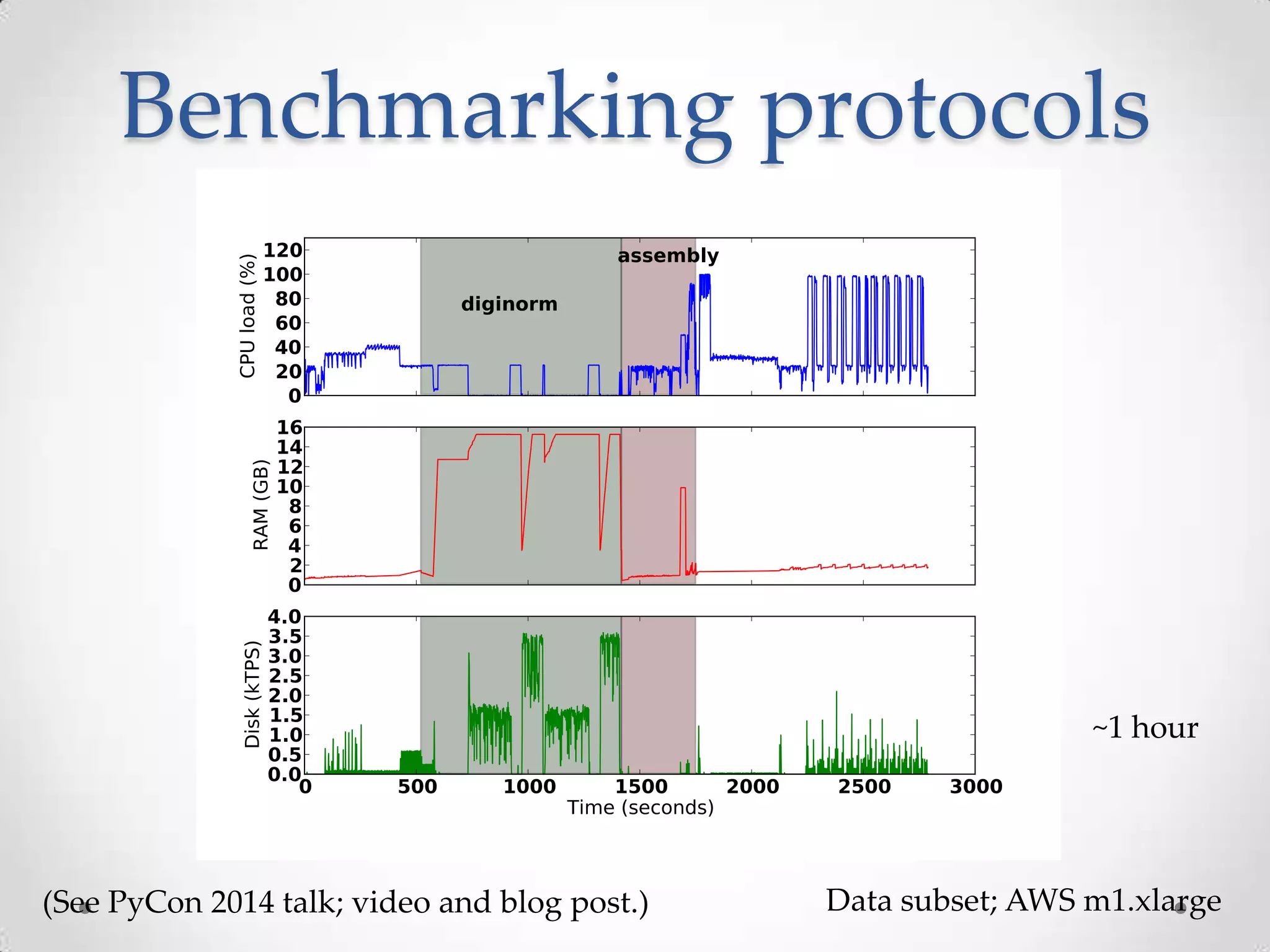
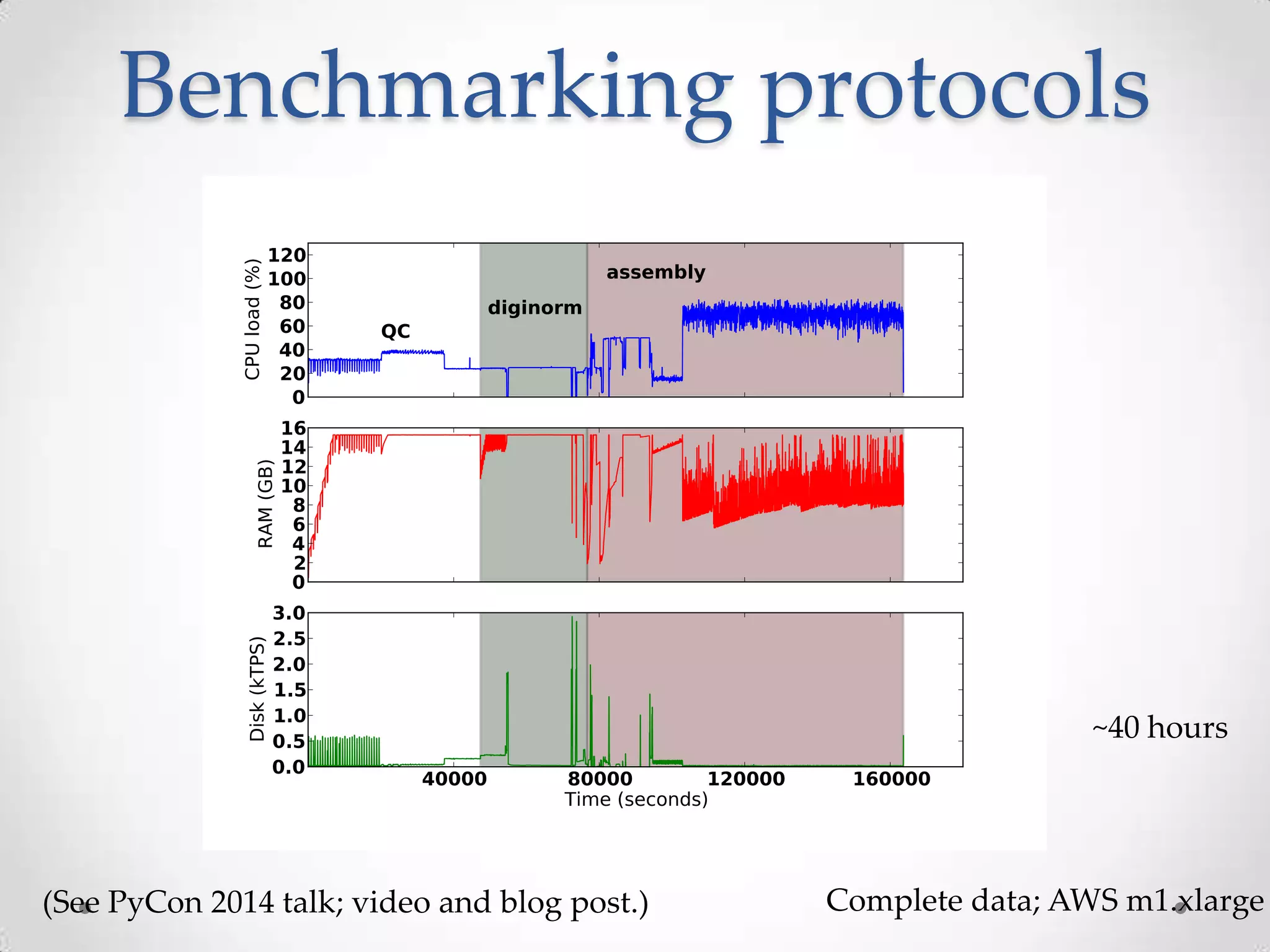
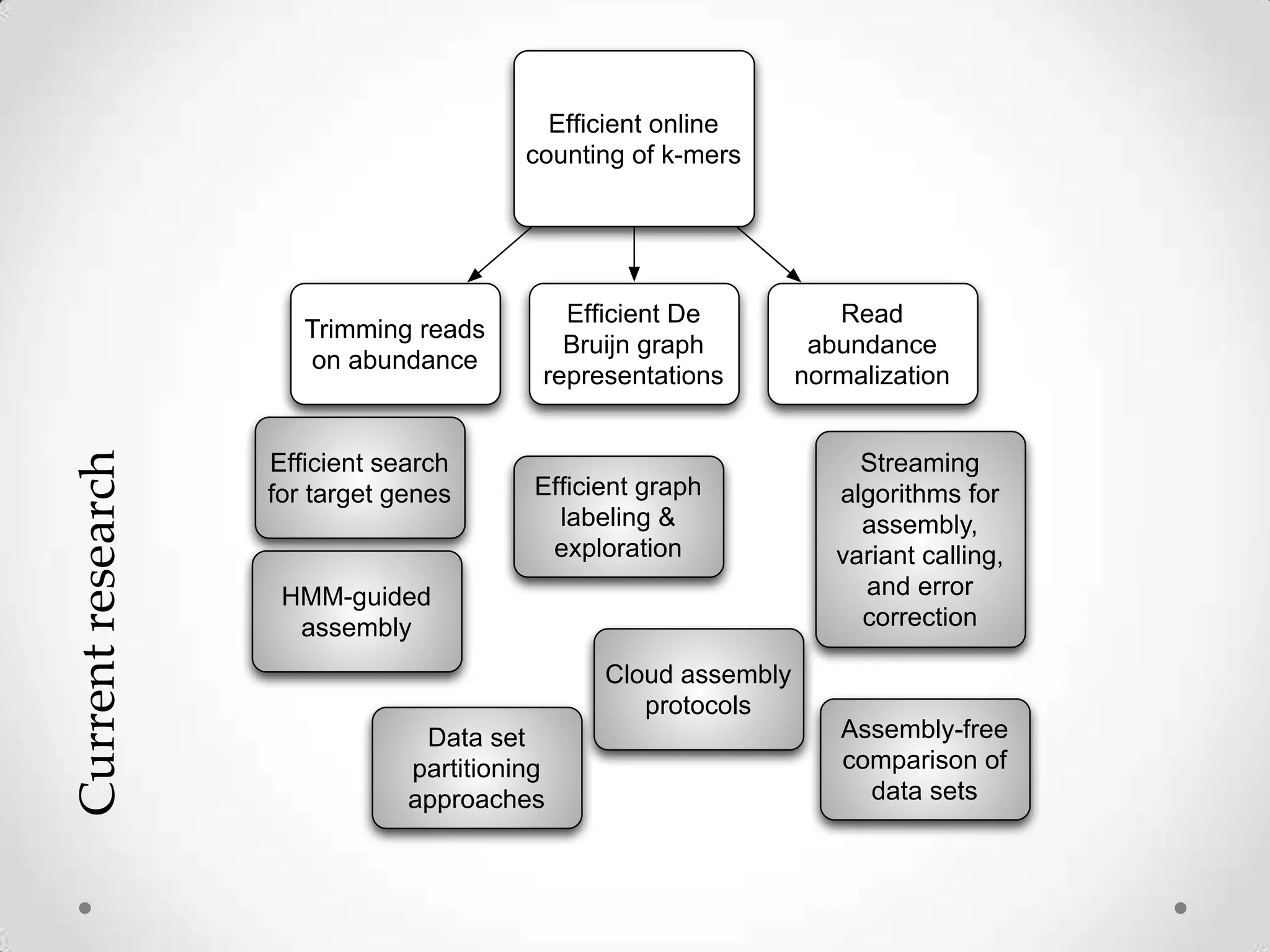
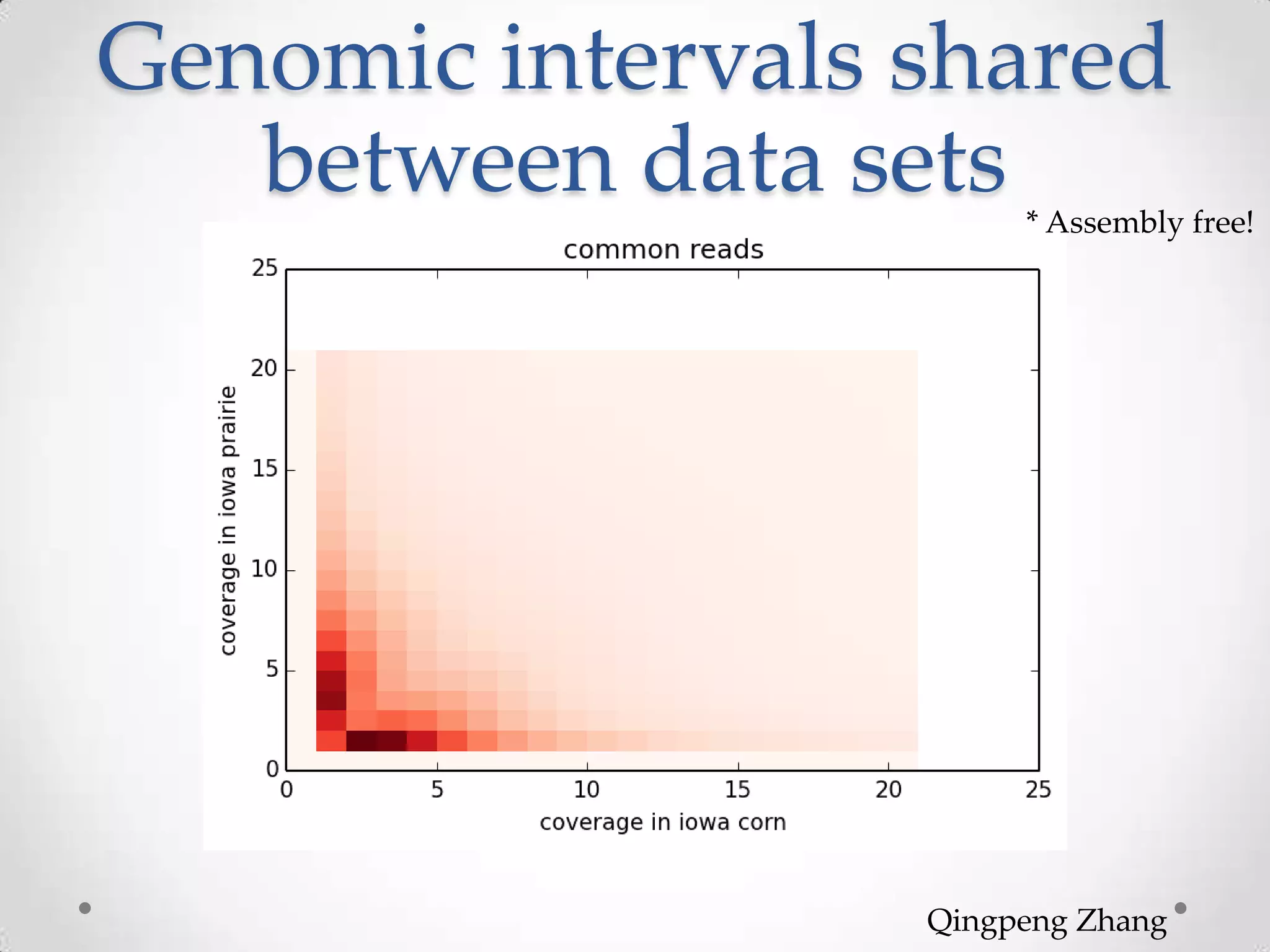
This document introduces khmer, a platform for scalable sequence analysis. It discusses how khmer uses k-mers to provide implicit read alignments and assemble sequences using de Bruijn graphs. It also describes some of the challenges with k-mers, such as each sequencing error resulting in novel k-mers. The document outlines khmer's data structures and algorithms for efficiently counting k-mers and represents de Bruijn graphs. It discusses how khmer has been applied to real biological problems and highlights areas of current research using khmer, such as error correction, variant calling, and assembly-free comparisons of data sets.

















































![[TDC 2013] Integre um grid de dados em memória na sua Arquitetura](https://cdn.slidesharecdn.com/ss_thumbnails/tdc2013integrandocoherencearqttv1-130526184010-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)



![Introducing BlackBerry 10 [Indonesian Version]](https://cdn.slidesharecdn.com/ss_thumbnails/blackberryclinique-introducingblackberry10-130303183033-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)





























































