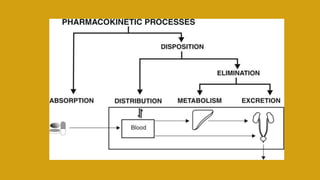
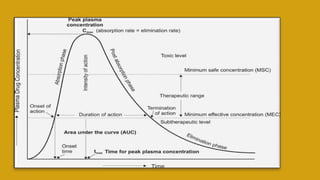
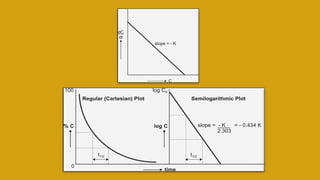
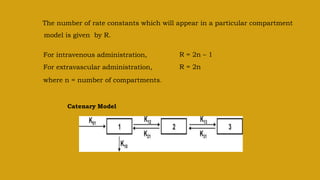
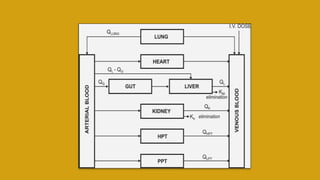
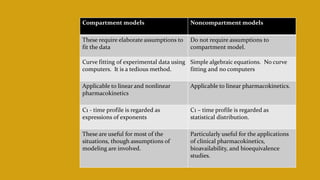
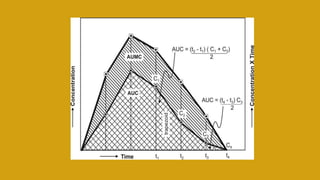
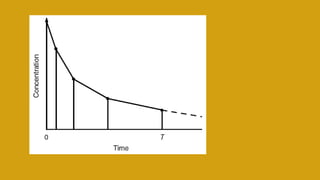
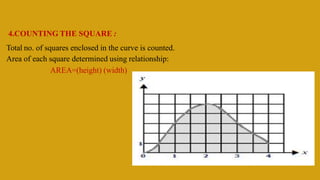
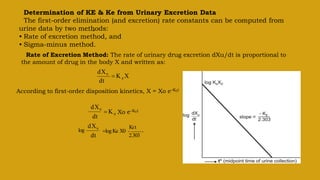
This document discusses pharmacokinetics and provides details about various pharmacokinetic parameters and models. It begins by defining pharmacokinetics as the study of the kinetics of drug absorption, distribution, metabolism and excretion. It then describes parameters that can be evaluated from plasma drug concentration-time profiles, including Cmax, Tmax, and AUC. Next, it discusses compartment models and physiological models that are used to analyze pharmacokinetic data and predict drug disposition. It concludes by covering the model-independent approach of noncompartmental analysis.