
Recommended
Recommended
More Related Content
What's hot
What's hot (20)
Similar to Introducción al laboratorio de microbiología
Similar to Introducción al laboratorio de microbiología (20)
Recently uploaded
Recently uploaded (13)
Introducción al laboratorio de microbiología
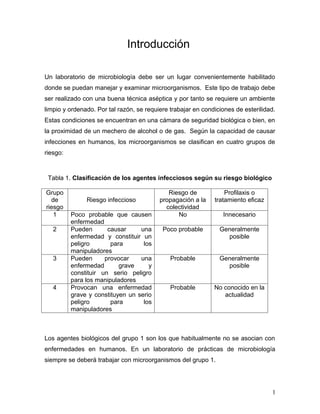
- 1. Introducción Un laboratorio de microbiología debe ser un lugar convenientemente habilitado donde se puedan manejar y examinar microorganismos. Este tipo de trabajo debe ser realizado con una buena técnica aséptica y por tanto se requiere un ambiente limpio y ordenado. Por tal razón, se requiere trabajar en condiciones de esterilidad. Estas condiciones se encuentran en una cámara de seguridad biológica o bien, en la proximidad de un mechero de alcohol o de gas. Según la capacidad de causar infecciones en humanos, los microorganismos se clasifican en cuatro grupos de riesgo: Tabla 1. Clasificación de los agentes infecciosos según su riesgo biológico Grupo Riesgo de Profilaxis o de Riesgo infeccioso propagación a la tratamiento eficaz riesgo colectividad 1 Poco probable que causen No Innecesario enfermedad 2 Pueden causar una Poco probable Generalmente enfermedad y constituir un posible peligro para los manipuladores 3 Pueden provocar una Probable Generalmente enfermedad grave y posible constituir un serio peligro para los manipuladores 4 Provocan una enfermedad Probable No conocido en la grave y constituyen un serio actualidad peligro para los manipuladores Los agentes biológicos del grupo 1 son los que habitualmente no se asocian con enfermedades en humanos. En un laboratorio de prácticas de microbiología siempre se deberá trabajar con microorganismos del grupo 1. 1
- 2. Normas generales de uso del laboratorio Para el desarrollo de las prácticas es conveniente tener en cuenta algunas normas elementales que deben ser observadas con toda responsabilidad: 1. Antes de realizar una práctica, debe leerse detenidamente el manual para adquirir una idea clara de su objetivo, fundamento y técnica (los resultados deben ser siempre anotados cuidadosamente apenas se obtengan). 2. Trabajar con calma y concentración. 3. Uso de bata en el laboratorio. 4. Lavarse las manos con agua y jabón al iniciar y finalizar el trabajo. 5. El orden y la limpieza son esenciales en todas las prácticas de laboratorio. Antes de comenzar se deberá desinfectar la superficie de trabajo. 6. Está prohibido comer, beber y fumar. Cuando se manipulan microorganismos hay que evitar llevarse las manos a la boca, nariz, ojos u oídos. 7. No se debe pipetear nunca con la boca. Utilizar siempre pipeteadores manuales o pipetas automáticas. 8. Los microorganismos deben manejarse siempre alrededor de la llama del mechero o en campana de seguridad biológica. 9. El transporte y almacenamiento de placas y recipientes que contienen cultivos representan un riesgo evidente. Colocar las placas y otros recipientes de forma segura para evitar que se caigan. 10. Nunca deben sustraerse cultivos de microorganismos del laboratorio. 11. Los desechos contaminados deberán ser esterilizados en el laboratorio antes de ser procesados como residuos. Todos los materiales de desecho que hayan entrado en contacto con microorganismos (como pipetas usadas, cajas petri o tubos), deben colocarse en bolsas de autoclave preparadas para tal efecto y proceder posteriormente a su esterilización. 12. Accidentespersonales, tales como derrame de reactivos, cortes y quemaduras, deben comunicarse inmediatamente al Instructor. 13. Los productos inflamables (gases, alcohol, éter, etc.) deben mantenerse alejados de las llamas de los mecheros. Si se manejan mecheros de gas se debe tener mucho cuidado de cerrar las llaves de paso al apagar la llama. 14. Cuando no se utilizan los mecheros, éstos deben guardarse y se debe estar seguro que se les ha apagado al final de cada práctica. 2
- 3. 15. Todo el material, especialmente los aparatos delicados, como lupas y microscopios, deben manejarse con cuidado evitando los golpes o el forzar sus mecanismos. 16. Los medios inoculados deben colocarse en las cámaras de cultivo con su identificación respectiva, ej.: número de mesa, nombre, naturaleza del espécimen o sustrato del cual se aísla. 17. Los tubos de ensayo que contengan medios de cultivos o cultivos de microorganismos, nunca deben abrirse en posición vertical, sino lo más horizontalmente posible (inclinados) y para quitar el tapón se mantendrán inclinados con una mano y se abrirán con la otra, que sostendrá a su vez el asa. Una vez abierto, se flamea por algunos segundos el orificio, repitiendo dicha operación una vez realizada la siembra (el tapón nunca debe dejarse sobre la mesa). 18. Las pipetas se tomarán de forma que sea el dedo índice el que tape su extremo superior para regular la caída de líquido. 19. Antes de utilizar las asas con hilos de platino que sirven para las siembras, éstas deben flamearse al rojo en posición vertical bajo la acción de la llama. Antes de efectuar la siembra debe esperarse algunos segundos a que se enfríen, pudiendo enfriarse también en el borde de la caja de Petri que contiene el medio de cultivo. Inmediatamente después de haberlas utilizado, deben flamearse nuevamente. 20. Al terminar cada práctica se procederá a limpiar cuidadosamente el material que se ha utilizado. Cada equipo se responsabilizará de su zona de trabajo y de su material. 3
- 4. PRACTICA 1: PREPARACION Y ESTERILIZACION DE MATERIAL INTRODUCCIÓN La presencia de microorganismos en todos los medios ambientales hace imprescindible que para estudiar una bacteria determinada sea necesario destruir todas aquellas que pudieran encontrarse contaminando los medios o los instrumentos de trabajo. Esto se consigue mediante la ESTERILIZACION, consistente en la destrucción de todo microorganismo. Generalmente en las prácticas de microbiología se utiliza el calor como método de esterilización rutinario, aunque existen otras técnicas. Los medios con contenido acuoso han de ser esterilizados en el AUTOCLAVE mediante calor húmedo, obtenido por calentamiento en un depósito hermético que contiene agua y del que previamente ha sido evacuado el aire con el fin de limitar las oxidaciones. En este sistema el vapor de agua sobrecalentado a 121 (2 atm) o 111 ºC (1.5 atm) difunde muy rápidamente, haciendo que los elementos termosensibles de las células se desnaturalicen, especialmente sus enzimas. Conceptos generales El concepto de esterilización implica la eliminación de todas las formas vivas. Según dicha definición, estéril significa libre de organismos. Otras metodologías, como desinfección, pasteurización, etc., conllevan una eliminación parcial de los microorganismos existentes. Métodos de esterilización: 1. Agentes físicos a) Calor - Calor seco: flameado, aire caliente (horno Pasteur) - Calor húmedo: vapor saturado (AUTOCLAVE). En el autoclave la esterilización se produce mediante vapor de agua a presión. En el autoclave se alcanza una temperatura de 121º C a una presión de 1 atmósfera sobre la presión ambiental. b) Filtración: permite la eliminación de los microorganismos de un medio líquido, sin la destrucción de estos. Para ello se hace pasar la muestra líquida a través de un filtro de membrana con tamaño de poro inferior al tamaño de los microorganismos (0,2-0,45 micras). Los microorganismos quedarán retenidos en el filtro y el fluido obtenido tras la filtración estará estéril. c) Radiaciones: - Rayos gamma: radiaciones ionizantes - Rayos Ultravioleta: de escasa penetración y de utilidad para eliminación de microorganismos de superficies. 4
- 5. Figura 1. Esquema de autoclave. 2. Agentes químicos: para esterilizar material (generalmente algunos tipos de plástico) que son termolábiles. Como el óxido de etileno y glutaldehído. 3. Otros métodos de eliminación: - calor: ebullición, pasteurización, tindalización. - agentes químicos: desinfectantes, antisépticos. OBJETIVOS: 1. Preparar material de vidrio de uso microbiológico para su esterilización en autoclave. 2. Emplear la técnica de vapor húmedo para el control de microorganismos. DESARROLLO DE LA PRÁCTICA MATERIAL Algodón Gasa Papel estrasa Tijeras Hilo de algodón Pinzas Cinta testigo Aguja enmangada 5
- 6. Pipetas de vidrio Cajas de Petri Mechero de alcohol Autoclave PROCEDIMIENTO PREPARACIÓN DEL MATERIAL A ESTERILIZAR 1. Colocar un filtro de algodón en el extremo posterior de las pipetas con ayuda de la aguja enmangada (con precaución de que el algodón no quede excesivamente compactado). El exceso de algodón externo será flameado con un mechero de alcohol. Las pipetas serán envueltas en papel estrasa cuidando de proteger la punta y posteriormente autoclavadas empaquetadas. Se debe tomar la precaución de orientar las pipetas, de forma que todas ellas se puedan extraer posteriormente sin ser expuestas a contaminación. 2. Cada una de las cajas de Petri (de vidrio) será envuelta en papel estrasa y el conjunto empaquetado para su esterilización en autoclave. 3. Se prepararán tapones con algodón y gasa para los matraces antes de ser esterilizados. 4. Los tubos con tapón de rosca conteniendo agua destilada (9 ml), serán esterilizados en autoclave, dejando ligeramente suelto el tapón. 5. Todo el material de vidrio será esterilizado en autoclave durante 15 minutos a 120 oC y 15 lb de presión. NOTA: El material de vidrio también se esteriliza mediante calor seco en el Horno Pasteur. Una vez limpio, el material se envuelve en aluminio o papel, se introduce en un contenedor (ej: pipetas en pipetero) y se mantiene en el horno durante dos horas a 160 ºC. En el caso de los portainóculos que se están utilizando constantemente, se utiliza el calor directo del mechero, que también sirve para flamear los bordes de tubos, matraces, pipetas, etc. RESULTADOS 1. Esquematizar el proceso de esterilización de material de vidrio. 2. Explicar el funcionamiento del autoclave. 3. Investigar los métodos físicos y químicos para el control de microorganismos, mencionando un uso específico para cada uno. CONCLUSIONES 6
- 7. PRACTICA 2. PREPARACION DE MEDIOS DE CULTIVO INTRODUCCION Un medio de cultivo es un conjunto de nutrientes, factores de crecimiento y otros componentes que crean las condiciones necesarias para el desarrollo de los microorganismos. La diversidad metabólica de estos organismos es enorme, por ello, la variedad de medios de cultivo es también amplia, y no existe un medio de cultivo universal adecuado para todos ellos. Constituyentes habituales de los medios de cultivo 1. Fuente de carbono y sales. En muchos casos, la glucosa, la lactosa u otras dextrosas se emplean como fuentes de carbono. Algunos medios de cultivo se complementan con sales como el NaCl o diversos fosfatos y/o sulfatos de potasio, magnesio, amonio, etc. 2. Agar. El agar se utiliza como agente gelificante para dar solidez a los medios de cultivo. el componente dominante en el agar bacteriológico es un polisacárido, al que acompañan algunas impurezas, que se obtiene de ciertas algas marinas (rodófitas). Un gel de agar al 1-2 % en agua licúa hacia los 100 oC y se gelifica alrededor de los 40 oC, dependiendo de su grado de pureza. Con la excepción de algunos microorganismos marinos, el agar no se emplea como nutriente. 3. Extractos. Para su preparación, cortos órganos o tejidos animales o vegetales (p. ej., carne, hígado, cerebro, semillas) son extraídos con agua y calor, y posteriormente concentrados hasta la forma final de pasta o polvo. estos preparados deshidratados se emplean con frecuencia en la confección de medios de cultivo. Son una fuente rica de alimentos, vitaminas y diversos factores de crecimiento. Ejemplo: extractos de carne, de levadura, de malta, etc. 4. Peptonas. Son mezclas complejas de compuestos nitrogenados y sales minerales que se obtienen por digestión enzimática o química de proteínas animales o vegetales (soja, carne, gelatina, caseína, etc.). Las peptonas son muy ricas en péptidos y aminoácidos, pero pueden ser deficientes en determinadas vitaminas y sales. 5. Fluidos corporales. sangre completa, sangre desfibrinada, plasma o suero sanguíneo son frecuentemente añadidos a los medios empleados para el cultivo de algunos patógenos. la sangre no puede esterilizarse y debe, por tanto, obtenerse en condiciones asépticas directamente de un animal sano. los fluidos corporales no sólo contribuyen como factores de crecimiento, si no también con sustancias que neutralizan inhibidores de crecimiento de algunas bacterias. 6. Sistemas amortiguadores (soluciones tamponadas). Algunos componentes son incorporados al medio al medio de cultivo para mantener el pH dentro 7
- 8. del rango óptimo del crecimiento bacteriano. Los microorganismos más comunes son neutrófilos (el pH óptimo para su crecimiento está próximo a la neutralidad), y sales como fosfatos bisódicos o bipotásicos, o sustancias como las peptonas, previenen variaciones del pH. 7. Indicadores de pH. indicadores ácido-base se añaden a menudo a los medios e cultivo para detectar variaciones del pH. 8. Agentes reductores. Cisteína, tioglicolato y otros son agentes reductores que se añaden a los medios de cultivo para crear condiciones que permitan el desarrollo de microorganismos microaerófilos o anaerobios. 9. Agentes selectivos. La adición de determinadas sustancias al medio de cultivo puede convertirlo en selectivo. Por ejemplo cristal violeta, sales biliares, azida sódica, telurito potásico, antibióticos, etc., a la concentración adecuada, actúan como agentes selectivos frente a determinados microorganismos. Tipos de medios de cultivo 1. Medios generales. Permiten el desarrollo de una gran variedad de microorganismos. 2. Medios de enriquecimiento. Favorecen el crecimiento de un determinado tipo de microorganismo, sin llegar a inhibir totalmente el crecimiento del resto. 3. Medios selectivos. Permiten el crecimiento de un tipo microbiano determinado, inhibiendo el desarrollo de los demás. 4. Medios diferenciales. Son aquellos en los que se ponen de relieve propiedades que posee un determinado tipo de microorganismo. OBJETIVOS: 1. Preparar medios de cultivo sólidos. 2. Esterilizar mediante calor húmedo los medios de cultivo. 3. Vaciar en placa los medios de cultivo. DESARROLLO DE LA PRÁCTICA 1. Preparación de medios de cultivo Caldo común. Composición por litro. Material: Extracto de carne 3g Peptona bacteriológica 10g Cloruro sódico 5g 8
- 9. Para prepararlo, se añaden los distintos componentes en un matraz de dos litros, se disuelven en agua, se ajusta el pH a 7.2-7.4 y se lleva al volumen final correspondiente. Posteriormente, se reparten en tubos utilizando un embudo al que se acopla una goma o un dosificador automático. Los recipientes se aíslan con tapones metálicos o de algodón envuelto en gasa y se esterilizan a 1 atm de presión. Agar nutritivo. - La composición es la misma que la del caldo común, pero se añade antes de esterilizar 15 g de agar por litro, disolviéndolo durante la esterilización. - Pesar y disolver los componentes en el medio en el volumen indicado de agua destilada. - Ajustar el pH del medio, si procede. - Para asegurar la completa disolución del agar, el medio se calienta hasta ebullición al mismo tiempo que se somete a agitación, previamente a su esterilización en el autoclave. Medio base para la fermentación de azúcares. Inicialmente se prepara un medio base pobre en aminoácidos de la siguiente composición por litro: Material: Extracto de carne 1g Peptona bacteriológica 10 g Cloruro sódico 5g Una vez disueltos los componentes, se ajusta el pH a 7 y se añaden 2 ml de solución de púrpura de bromocresol (PBC: 1.6 g en 100 ml de etanol al 45%. Se disuelve inicialmente en etanol al 95 y posteriormente se diluye con agua) por litro. Este es un indicador de pH que vira a amarillo por debajo de pH 5.2, por lo que permite la detección de la producción de ácido en el medio. El medio se reparte en tres volúmenes iguales a los que se añade 10 g por litro de glucosa, lactosa o sacarosa. Una vez disuelto, se reparte en tubos en los que previamente se ha introducido una campana Durham, pequeño tubo de ensayo que se dispone en posición invertida para detectar la presencia de gases. El medio ha de ocupar completamente el interior de estas campanas para poder detectar posteriormente el desprendimiento de gases en la fermentación. (No olvidar marcar cada tubo con la inicial del azúcar que contenga). Una vez taponados, se esterilizan a 0.5 atm durante 20 min. 2. Esterilización de medios de cultivo 9
- 10. Una vez preparado el medio, éste se debe esterilizar lo antes posible para evitar el crecimiento de microorganismos: - Medios sólidos en placa: Tapar el matraz con tapón de algodón y cubrir con papel de aluminio para llevar a esterilizar en el autoclave (121º C, 15-20 min). Una vez estéril repartir el medio en placas de Petri estériles y dejar en reposo para su solidificación. - Medios sólidos en tubo (agar inclinado): Tras la ebullición del medio con agar, éste se reparte en los tubos de cristal, de forma que queden llenos hasta aproximadamente la mitad. Los tubos se cubren con tapón (de aluminio) para llevar al autoclave. Una vez esterilizado, los tubos se disponen en posición inclinada para su solidificación. - Medios líquidos: Una vez disueltos los componentes del medio en el matraz, repartir en su caso en tubos de 2-4 ml por tubo, cubrir con tapón y llevar a esterilizar en el autoclave. Alternativamente, se puede esterilizar el medio en el matraz, y posteriormente repartirlo con la ayuda de pipetas estériles en tubos de plástico estériles. - Medios semisólidos: Se preparan con concentraciones inferiores de agente solidificante (agar-agar). El medio se reparte en tubos. RESULTADOS 1. Esquematizar el proceso de preparación de medios de cultivo. 2. Investigar tipos de medios de cultivo, su composición y uso. 3. Mencione las características que debe tener un medio de cultivo para el crecimiento de un fitopatógeno. 4. Indique como determinaría la necesidad de nutrientes esenciales de un microorganismo. CONCLUSIONES 10
- 11. PRACTICA 2. AISLAMIENTO DE MICROORGANISMOS INTRODUCCIÓN En la naturaleza los microorganismos se encuentran en comunidades más o menos complejas. Por ello, una técnica especial en microbiología es la que permite la obtención de cultivos puros o axénicos, a partir de los cuales son posibles estudios sobre las propiedades de un microorganismo concreto, ya que un cultivo puro es aquel que contiene un solo tipo de microorganismo. Aunque existen numerosas técnicas de aislamiento, las más utilizadas emplean un medio sólido sobre el que se desarrollarán las colonias macroscópicas como resultado del crecimiento y división de los microorganismos. Teóricamente cada colonia procede de la multiplicación de una sola célula; sin embargo, una colonia también puede ser el resultado de la multiplicación de un agregado de células. Por otra parte, no todas las células bacterianas son capaces de formar colonias, ya que no crecen en medios de cultivo. Por tal razón, lo más correcto es hablar de unidades formadoras de colonias (UFC) al referirnos al origen de una colonia. Manejo de las muestras y toma del inoculo La toma del inóculo de una muestra de un medio sólido o líquido es simple, pero requiere atención. Se recomienda lo siguiente: 1. Coloque frente al manipulador el mechero y la preparación o muestra que contiene los microorganismos, así como el resto del material necesario (tubos, placas, pipetas, etc.), que pueda ser alcanzado con facilidad. 2. Tome el asa de siembra y flamee el filamento hasta que este alcance un rojo incandescente. Enfríelo en la proximidad de la llama (unos 10 s). Recuerde que las llamas “frías” son anaranjadas y no esterilizan. Las llamas muy “calientes” son de color azul, y si esterilizan. 3. Tome con la otra mano el recipiente (tubo, placa, etc.) que contiene la muestra. Si la muestra está en un tubo, quite el tapón con los dedos meñique y anular como se muestra en la figura 2 y flamee la boca del tubo. Si la muestra está en una placa de Petri, coloque la placa invertida sobre la mesa de trabajo y levante la parte que contiene el medio de cultivo con los microorganismos. Llévela a la proximidad de la llama del mechero. 4. Trabajando en todo momento en la proximidad de la llama, introduzca el asa de siembra y tome una pequeña muestra del cultivo. Si el medio es liquido, agite ligeramente el tubo y tome una muestra que quedará adherida por tensión superficial en el extremo del filamento del asa de siembra. Si los microorganismos están en forma de colonias o sobre un medio sólido, tome 11
- 12. una pequeña porción de una de las colonias mediante un ligero roce con el asa de siembra. 5. Deposite el inóculo en un medio de cultivo estéril. 6. Siembre sobre la superficie del medio de cultivo. Marque las placas con la identificación del cultivo, la fecha y el nombre. Incube en las condiciones adecuadas durante el tiempo necesario para que se desarrollen los microorganismos. 7. Antes de dejar el asa de siembra sobre la mesa, flaméela de nuevo con el objeto de esterilizarla. 12
- 13. Figura 2. Manejo de muestras y toma del inóculo. OBJETIVOS 1. Aislar un microorganismo de una mezcla de varios (suelo, vegetales y alimentos en descomposición, plantas enfermas, aguas grises, etc.). 2. Obtención de cultivos puros de microorganismos a partir de un cultivo mixto, mediante estría en medio sólido. 3. Recuento de microorganismos empleando la técnica de diluciones seriadas. Procedimiento a) Aislamiento mediante agotamiento por estrías. Se trata de un método rápido y simple de agotamiento progresivo y continuo del inóculo sobre un medio sólido contenido en una placa de Petri. El objetivo final consiste en obtener un número reducido de colonias distribuidas individualmente sobre la superficie de la placa. Al incubar esta, cada una de las bacterias originará una colonia. Normalmente, una colonia aislada es un cultivo puro. Material Muestra mixta con microorganismos (suelo, basura, agua, alimentos en descomposición, yoghurt, etc.). Medio de cultivo (Agar nutritivo) en placas de Petri Tubos de ensaye Agua destilada estéril Mechero Bunsen Mechero de alcohol Pipeta de vidrio estéril Asa de siembra 1. En condiciones estériles (mesa desinfectada con benzal y mechero encendido), resuspender 0,1-1 g de muestra en un tubo con 1 ml de agua destilada estéril. 2. Agitar bien en vórtex. Dejar sedimentar las partículas. 3. Tomar una muestra del sobrenadante con el asa de platino (como fue indicado anteriormente), o con pipeta de 2 ml estéril, y extender con el asa (previamente flameada) en una placa de Agar Nutritivo (ver figura 3). 13
- 14. 4. Una vez sembrada la muestra por estriado, marcar la caja con la clave asignada y número de aislamiento que le corresponde (por ejemplo, A1). Incubar a 28 oC durante 24 h, o hasta que haya crecimiento de microorganismos. Figura 3. Aislamiento de microorganismos a partir de una muestra mixta. 14
- 15. Figura 4. Técnica de aislamiento por estría en superficie. RESULTADOS 1. Esquematice el proceso realizado para el aislamiento de microorganismos a partir de una muestra mixta. 2. Reporte en un cuadro el número de colonias obtenidas y su morfología macroscópica. Colonia Forma Borde Elevación Superficie Fotografía 1 2 3 3. Indique cuales fueron las colonias seleccionadas para resiembra y obtención de un cultivo puro. 15
- 16. b) Obtención de un cultivo puro a partir de un cultivo mixto. El trabajo con microorganismos no se realiza con células aisladas, sino con poblaciones extensas y homogéneas del microorganismo a estudiar. Por lo tanto, es necesario utilizar técnicas que permitan obtener un cultivo puro, y luego cultivar a gran escala dicho microorganismo. Para obtener cultivos puros se utilizarán instrumentos estériles y colonias aisladas del microorganismo deseado. Para aislar un microorganismo de una mezcla de varios y obtener un cultivo puro del mismo, la técnica más utilizada es la de siembra por estría en placa Petri, en medio sólido, que permite obtener colonias separadas de individuos que proceden por división de una única célula. Estas colonias nos sirven para iniciar un cultivo a gran escala. Otra técnica consiste en obtener una suspensión de la mezcla de microorganismos y hacer diluciones seriadas que se vierten en una placa Petri hasta conseguir colonias aisladas. Una vez que se ha obtenido un cultivo puro, se puede mantener haciendo resiembras en tubos de agar inclinado o bien congelando las células en glicerol (10-30%) a -80°C, en nitrógeno líquido a -173°C, o mediante liofilización. Material Cultivos de microorganismos en agar nutritivo Medio de cultivo (Agar nutritivo) en placas de Petri Mechero Bunsen Mechero de alcohol Asa de siembra Estereoscopio 1. Observación de colonias aisladas. Con el uso de un estereoscopio se observará la morfología de cada una de las colonias desarrolladas en las placas de Petri (aislamiento 1). 2. Resiembra. Tomar una muestra del cultivo mixto, con ayuda del asa de siembras previamente flameada. Debe flamearse el asa antes y después de 16
- 17. tomar la muestra. La muestra se extiende con suavidad con el asa sobre la superficie del medio de agar nutritivo en placa Petri (ver figura 4). Una vez finalizada la resiembra, se vuelve a tapar la placa y está se dispone en posición invertida para llevar a incubar a 28 ºC durante 24 horas. 3. Se harán las resiembras necesarias hasta obtener cultivos puros de las colonias seleccionadas. RESULTADOS 1. Reporte en un cuadro el número de colonias obtenidas y su morfología macroscópica. Colonia Forma Borde Elevación Superficie Fotografía 1 2 3 2. Indique cuales fueron las colonias seleccionadas para resiembra y obtención de un cultivo puro. 3. ¿Cómo demuestra que tiene un cultivo puro aislado? c) Recuento de microorganismos. Frecuentemente, las muestras naturales tienen un número muy elevado de microorganismos, por lo que se requiere diluirlas para poder efectuar recuentos. Para ello, se realizan diluciones seriadas que posteriormente se siembran en placas con el medio de cultivo adecuado. El método más habitual para la determinación de células viables se basa en contar el número de colonias. Según el concepto de UFC, solo se van a cuantificar los microorganismos que se multipliquen en las condiciones concretas del ensayo (temperatura, medio de cultivo, atmósfera, etc.). La inoculación de la placa con la muestra que contiene 17
- 18. los microorganismos puede realizarse: a) mezclándolo previamente con el medio fundido y temperado, para verterlo después sobre la placa de Petri vacía (método de vertido en placa o siembra en profundidad); b) directamente sobre el medio ya solidificado en la placa (método de extensión sobre placa). Material 5 tubos con 9 ml de solución salina o agua destilada estéril Pipetas estériles de 1 ml Placas de agar nutritivo Muestra: resuspender una colonia en 10 ml de agua destilada estéril 1. Diluciones seriadas (Ver figura 5) Abrir el recipiente que contiene la muestra problema y flamear la boca del mismo. Tomar 1 ml de la muestra problema. Flamear de nuevo la boca del recipiente y taparlo. Transferir 1 ml al primero de los tubos con 9 ml de agua destilada estéril o solución salina estéril, flameando la boca del tubo después de retirar el tapón y antes de reponerlo. Agitar el tubo para homogenizar totalmente la suspensión. De esta forma, se consigue diluir la muestra inicial 10 veces (dilución 10-1). Agitar bien el tubo con la primera dilución. Con una nueva pipeta estéril de 1 ml, transferir 1 ml de esta primera dilución al segundo tubo con 9 ml de agua. Agitar el tubo y marcarlo como dilución 10-2. Repetir la misma operación para conseguir las diluciones 10 -3, 10-4, 10-5, etc. El número de diluciones dependerá de la carga microbiana que se sospeche que contiene la muestra. 2. Siembra en profundidad (Ver figura 5) Transferir con una pipeta estéril 1 ml de cada dilución de la muestra a placas de Petri con el medio de agar fundido y atemperado a 45-50 oC. Mezclar y distribuir uniformemente (girando la placa con suavidad), se deja enfriar hasta su solidificación. Invertir las placas e incubar en las condiciones adecuadas requeridas. 3. Extensión sobre placa (Ver figura 6) Transferir con una pipeta estéril 0.1 ml de cada dilución de la muestra a placas de Petri con el medio de cultivo ya solidificado. Este inóculo debe extenderse de forma homogénea por toda la superficie de la placa, para lo cual se utilizará la varilla de vidrio acodada, previamente esterilizada (impregnándola de alcohol y pasándola por la llama del mechero). incubar las placas en posición invertida a 30 oC durante 24 horas. 18
- 19. Después del periodo de incubación examinar las placas. Las colonias aparecerán sobre la superficie del agar. Cada colonia representa los descendientes de una unidad formadora de colonia (UFC). A B Figura 5. Recuento de microorganismos. A. Diluciones seriadas B. Siembra en profundidad. Serie de disoluciones conocidas de la muestra cayado 19 Siembra por Extensión en Superficie
- 20. Figura 6. Siembra por extensión en placa. Resultados 1. Calcular el número de UFC/ml de las muestras problema a partir del recuento de colonias. 2. Discutir las condiciones necesarias para efectuar un análisis adecuado de recuento de colonias. 3. ¿Se obtendría el mismo resultado si cuantificáramos las bacterias presentes en una muestra por turbidimetría o por diluciones seriadas? ¿Por qué? 4. Aplicando la técnica de esta práctica, considere que se han sembrado 2 placas de medio de cultivo a partir de las diluciones 10 -4 y 10-5. Tras la incubación de las placas a 37 oC durante 24 horas, no apareció ninguna colonia. ¿Significa esto que no había ninguna bacteria en la muestra problema? ¿Por qué? 5. Considerando, que para un cultivo se toman 2.5 g de tierra y se resuspenden en solución salina estéril hasta un volumen final de 50 ml. A continuación, se hacen diluciones decimales y se siembran 0.2 ml de cada una sobre un medio adecuado. Tras la incubación, en la placa de la dilución 10-6 se desarrollan 200 colonias. Calcule el número de UFC por gramo de tierra. 6. Investigar otras técnicas de recuento bacteriano. CONCLUSIONES PRÁCTICA 4. OBSERVACION MICROSCOPICA DE MICROORGANISMOS INTRODUCCION La gran mayoría de los organismos unicelulares son invisibles para el ojo humano, por lo que e uso del microscopio resulta indispensable para observar a estos 20
- 21. seres vivos. Para lograr una buena observación microscópica, además de disponer de un adecuado aumento de la imagen, es necesario tener en cuenta dos factores más: el contraste y la resolución. Los objetos deben tener un cierto grado de contraste con el medio circundante para poder ser percibidos a través del microscopio. Por otra parte, el grado o poder de resolución (la capacidad para ver separados dos objetos muy próximos entre si) depende de las características del sistema de lentes que posea el microscopio y está limitado por la longitud de onda de la luz empleada, el índice de refracción del medio en que se realiza la visualización y la apertura numérica del objetivo. El microscopio óptico compuesto (ver figura 7) con iluminación en campo claro es el más utilizado en los laboratorios de microbiología. El microscopio compuesto recibe este nombre porque su sistema óptico dispone de dos o más lentes de aumento. Estructuralmente, un microscopio se divide en dos partes: el soporte y el sistema óptico. Figura 7. Microscopio óptico compuesto. 1. Soporte. Es elemento que proporciona un apoyo fijo y estable a todo el resto de elementos del microscopio. Sus partes principales son las siguientes: a) La base, que normalmente alberga la fuente de iluminación (lámpara de incandescente o halógena). Muchos microscopios poseen en su base un reóstato para variar la intensidad de la luz que llega a la preparación. En algunos modelos la base incorpora un sistema portafiltros con varios filtros de luz y un diafragma del campo luminoso. 21
- 22. b) El brazo soporta el sistema óptico, el cabezal portaoculares (mono o binocular) y el revolver portaobjetitos. En el brazo se dispone también el mecanismo de anclaje de la platina, dotado de un sistema de movimiento en el eje vertical controlado por los tornillos macrométrico y micrométrico. c) La platina es la pieza donde se coloca la preparación microscópica para su observación. Presenta un orificio central por donde pasa la luz y esta equipada con un sistema de fijación de la preparación, que permite el desplazamiento del portaobjetos sobre la platina en los dos ejes del plano horizontal y facilita el cambio del campo de observación. d) Los tornillos macrométrico y micrométrico controlan el movimiento de la platina en el eje vertical y así permiten el enfoque de la preparación. El primero se emplea para un enfoque rápido cuando se trabaja con el objetivo de menor aumento (x 10), mientras que el tornillo micrométrico permite afinar el enfoque cuando se utilizan los objetivos de mayor aumento (x40, x100). 2. Sistema óptico. El sistema óptico de un microscopio compuesto consta normalmente de tres lentes o conjuntos de lentes denominados condensador, objetivo y ocular. Las dos últimas se alojan en los extremos del tubo del microscopio y su acción combinada produce el aumento total de la imagen, puesto que son las lentes que se interponen entre la preparación el ojo del observador. a) El condensador, es la lente o conjunto de lentes colocadas entre la fuente de iluminación y la preparación. Su misión es recoger los rayos de luz procedentes de la fuente de iluminación y concentrarlos sobre la preparación. El condensador incorpora un diafragma tipo iris, que permite ajustar la cantidad de luz que llega a la preparación y actúa de un modo muy eficaz sobre el contraste final de de la imagen microscópica. Cuando se empleen grandes aumentos, es fundamental conseguir una iluminación óptima; en este caso es necesario realizar un centrado tanto de la fuente de luz como del condensador, además de un correcto diafragmado. b) El objetivo genera una imagen ampliada e invertida que se forma en el plano focal anterior del ocular. Los objetivos están montados sobre un soporte rotatorio o revólver que posibilita cambiar de un objetivo a otro mediante un sencillo giro. A diferencia del resto de objetivos, el objetivo de inmersión (normalmente de 100 aumentos), está diseñado para emplearse en contacto directo con aceite de inmersión para microscopia, que se aplica previamente sobre la preparación y se interpone entre esta y el objetivo. Como los índices de refracción del portaobjetos (vidrio) y del aceite son prácticamente iguales, los rayos de luz no son refractados al pasar de un medio a otro, lo que se traduce en una mayor resolución. 22
- 23. c) El ocular es la lente, o conjunto de lentes que aumentan a modo de lupa la imagen real proyectada por el objetivo y permite que sea percibida en la orientación y posición correctas por el ojo del observador. El ocular es el elemento del sistema óptico que se encuentra más próximo al ojo del observador. Suele llevar grabado el número de aumentos (x10, generalmente). OBJETIVOS 1. Conocer el manejo y uso del microscopio óptico. 2. Practicar la observación con el microscopio y familiarizarse con el uso del aceite de inmersión. 3. Identificar cultivos puros por observación microscópica. 4. Observar microorganismos vivos e identificar su morfología en condiciones naturales con ayuda del microscopio. 5. Preparar frotis bacterianos y realizar tinciones simples empleando dos tipos de colorantes. 6. Observar la morfología bacteriana y distinguir los distintos tipos de agrupaciones. 7. Diferenciar entre bacterias Grampositivas y gramnegativas. 8. observar las formas y tamaños de las endosporas en varias especies bacterianas formadoras de endosporas. . PROCEDIMIENTO a) Manejo y uso del microscopio Material Microscopio óptico por equipo Portaobjetos Cubreobjetos Algodón 1. Compruebe que las lentes del ocular y de los objetivos están limpias. De no ser así, límpielas cuidadosamente con un algodón o papel especial para óptica. No toque las lentes con los dedos. 2. Coloque el portaobjetos (sin cubreobjetos y con la muestra en la parte superior) sobre la platina, de manera que quede asegurado por las pinzas de sujeción. Compruebe previamente que el objetivo de menor aumento (x4 o x10) está en posición de empleo. 23
- 24. 3. Disminuya la iluminación de la preparación bajando el condensador (más contraste), y con el diafragma abierto. 4. Coloque en posición el objetivo de 10 aumentos (x10) y realice la operación de enfoque de la manera siguiente: a) Sin mirar por el ocular, gire el tornillo macrométrico y acerque al máximo la lente del objetivo a la preparación. b) Mirando por el ocular, gire el tornillo macrométrico en sentido opuesto y aleje el objetivo de la preparación lentamente hasta que obtenga un enfoque nítido. 5. Gire el revólver y coloque en posición de objetivo X40. Suba ligeramente el condensador hasta obtener una iluminación suficiente para ese aumento. La imagen debería estar casi enfocada, pero puede ser necesario afinar el enfoque girando un poco el tornillo micrométrico. La distancia óptima funcional del objetivo de X40 es de unos pocos milímetros por encima de la preparación, por lo que podría romperse si no se maneja con precaución. Por otra parte, cuando se ha realizado una observación con el objetivo de inmersión (X100), no puede volver a enfocar la muestra con el objetivo de X40, ya que este lente no debe nunca estar en contacto con el aceite. 6. Empleo del objetivo de inmersión (X100): a) Una vez que haya enfocado con el objetivo de X40, gire el revólver hacia el objetivo de inmersión sin llegar a colocarlo en posición. b) Coloque una gota de aceite de inmersión sobre la muestra en la zona de observación. c) Gire con precaución el revólver hasta situar en posición el objetivo de inmersión asegurándose que este no toca la preparación, pero si la gota de aceite. d) Gire ligeramente el tornillo micrométrico hasta conseguir un enfoque nítido. La preparación debería estar prácticamente enfocada si se ha realizado un enfoque adecuado con el objetivo de X40. De no ser así, es preferible volver a enfocar de nuevo un campo distinto, libre de aceite de inmersión. e) Finalizada la observación de una preparación y antes de retirarla de la platina, se colocará el objetivo de menor aumento. Nunca retire la preparación con el objetivo de inmersión en posición de observación. f) Retire la preparación y limpie el objetivo de inmersión con cuidado empleando algodón. Compruebe también que el objetivo de X40 está perfectamente limpio. RESULTADOS 1. Investigar el funcionamiento del sistema óptico del microscopio compuesto. 24
- 25. 2. investigar el funcionamiento y uso de otros microscopios ópticos especiales. b) Examen en fresco de microorganismos El examen en fresco de una suspensión microbiana es la técnica de observación microscópica más fácil y rápida de realizar. Es el método de elección cuando se desea evitar la alteración que sufren los microorganismos de una muestra cuando esta se somete a una fijación y a una posterior tinción. El examen en fresco posibilita la observación de microorganismos vivos, y permite estudiar determinadas características como su morfología y tamaño reales, su color natural o su movimiento. Material Mechero Bunsen Asa de siembra Portaobjetos y cubreobjetos Muestras obtenidas en aislamiento (práctica anterior) Muestra de agua estancada Microscopio de campo claro Aceite de inmersión 1. Usando un gotero o una pipeta, colocar una gota de agua sobre un portaobjetos bien limpio y desengrasado. 2. Usando el asa de siembra previamente flameada, tomar una colonia o una porción de esta (si se encuentra en un medio sólido), y extender suavemente sobre la gota de agua en el portaobjetos hasta ocupar una superficie de aproximadamente 1 cm2 . 3. Colocar el cubreobjetos sobre el portaobjetos. Para evitar que se formen burbujas, depositar el cubreobjetos sobre la gota, primero una arista y luego, lentamente, el resto. 4. Observar a través del microscopio. 25
- 26. NOTA: Tenga en cuenta que el calor generado por el foco luminoso irá secando la preparación dificultando la observación de la muestra. Para que esto no suceda, como alternativa se puede aplicar parafina en los bordes del cubreobjetos. RESULTADOS 1. Identifique si existe movimiento activo de los microorganismos (por medio de cilios o flagelos, o por deslizamiento), o movimiento pasivo de vibración o “browniano”. 2. Identifique (con dibujos o fotografías) los diferentes tipos morfológicos de los microorganismos observados. 3. Señale las ventajas e inconvenientes del examen en fresco. c) Preparación de un frotis bacteriano La mayoría de las bacterias son transparentes, por lo que suele ser necesario recurrir al uso de tinciones si se desea observar a estos seres vivos con un microscopio óptico convencional. Las tinciones aumentan artificialmente el contraste entre las células bacterianas y el medio circundante. La tinción que hace uso de un solo colorante se denomina tinción simple. Las tinciones más usadas en microbiología requieren que la muestra en estudio se someta al proceso de fijación. Habitualmente, las muestras bacterianas se preparan para ser observadas en forma de frotis o extensión sobre portaobjetos, lo que asegura una adecuada 26
- 27. separación entre las células y facilita que la tinción posterior se distribuya de manera homogénea. Para hacer un frotis o extensión, la suspensión bacteriana se homogeneiza bien, se extiende sobre un portaobjetos y se fija (Figura 8). Material Mechero Bunsen Asa de siembra Portaobjetos Cultivos bacterianos 1. Coloque sobre un portaobjetos limpio una pequeña gota de agua. La cantidad de agua no debe superar la que se puede reunir tomando dos veces con el asa de siembra. 2. a) Transfiera una pequeña cantidad del cultivo (la equivalente a una pequeña porción de una colonia) a la gota. Empleando el asa de siembra previamente flameada, disperse los grumos y luego remueva la gota mediante movimientos circulares hasta formar una suspensión homogénea. Extienda la preparación para facilitar el secado. 2. b) Si parte de un cultivo bacteriano en medio liquido, coloque una gota pequeña de la suspensión bacteriana (tomando muestra varias veces con el asa de siembra) sobre el portaobjetos. Extienda la preparación para facilitar su secado. 3. Fijación de las bacterias al vidrio portaobjetos: a) Por calor. Caliente el portaobjetos a la llama con rápidos pases sobre el mechero Bunsen. No utilice pinzas, el calor en exceso dañaría la integridad-estructura de las células. b) Con metanol (apropiado para bacterias procedentes de medio liquido). Añadir unas gotas de metanol sobre la extensión previamente desecada al aire. Retirar de inmediato el exceso de metanol del portaobjetos, golpeándolo ligeramente por una orilla sobre un papel de filtro. Esperar a que el metanol se evapore completamente. 27
- 28. Figura 8. Preparación de un frotis bacteriano. d) Tinción simple 28
- 29. Como la mayoría de los microorganismos carecen de una coloración natural, hay que teñirlos previamente para hacerlos resaltar de manera artificial del fondo de la imagen. Una forma sencilla de conseguir este contraste consiste en realizar tinciones simples con un colorante. Para poner de manifiesto o realzar de manera específica determinadas características morfológicas o estructurales de las células microbianas se emplean tinciones diferenciales con varios colorantes. Estas últimas tienen un gran interés para identificar y clasificar las bacterias. La tinción diferencial más utilizada en bacteriología es la tinción de Gram. Otras tinciones de gran importancia bacteriología son las que sirven para teñir capsulas mediante tinción negativa, las que se emplean para diferenciar las bacterias aaaaa2acido alcohol-resistentes” y las que permiten poner de manifiesto estructuras bacterianas características como las endosporas. Material Mechero Bunsen Asa de siembra Portaobjetos Cultivos bacterianos Colorantes para tinción: a) Solución de cristal violeta al 1% b) Solución de safranina al 0.5 % Microscopio Aceite de inmersión 1. Cubrir un frotis con cristal violeta y el otro con safranina cuidando que no quede ninguna porción sin teñir. deje actuar a los colorantes durante 1 minuto. 2. lavar las preparaciones con agua para eliminar el exceso de colorante. para ello incline el portaobjetos y aplique el agua corriente de grifo en su parte superior de manera que resbale poco a poco sobre l frotis. No dirija el agua directamente sobre el frotis pues podría despegar parte de la preparación. 3. Secar el portaobjetos por simple presión entre dos papeles de filtro o mediante calor. En ningún caso frote el portaobjetos con papel u otro tejido. 4. Observar la preparación al microscopio con el objetivo de inmersión. 29 Figura 9. Tinción simple.
- 30. Figura 10. Morfología bacteriana y tipos de agrupaciones. RESULTADOS 1. Dibujar o fotografiar la morfología y agrupación de las bacterias observadas. 2. ¿Observó microorganismos diferentes a las bacterias? ¿Qué características morfológicas presentaron? 3. Indique que etapas debería seguir para preparar una extensión de las siguientes muestras: yogur, saliva, sedimento de un río, una colonia bacteriana resuspendida en agua. 4. ¿Qué finalidad tiene fijar las muestras cuando se realiza un frotis bacteriano? 5. Señale las ventajas y desventajas de la tinción simple respecto del examen en fresco. 6. Con los resultados de una tinción simple, ¿se puede saber si la muestra teñida es un cultivo puro? 30
- 31. e) Tinción diferencial de Gram Las tinciones diferenciales se denominan así porque son capaces de diferenciar estructuras o incluso microorganismos que poseen características superficiales distintas. Este tipo de tinciones requieren de más de un colorante. La tinción diferencial más empleada es la tinción de Gram, llamada así en honor de Christian Gram, patólogo danés que la describió por primera vez en 1884. La aplicación de esta tinción permite clasificar las bacterias en dos grandes grupos: las bacterias Grampositivas y las bacterias gramnegativas. Estos dos grupos poseen características estructurales superficiales notablemente distintas (Figura 11). El fundamento radica en la diferente estructura de la pared celular de ambos grupos: las bacterias Gram+ tienen una gruesa capa de peptidoglicano en su pared, mientras que las bacterias Gram- tienen una capa de peptidoglicano más fina y una capa lipopolisacarídica externa. Tras la tinción con el primer colorante (Cristal violeta) se efectúa un lavado con etanol que arrastrará al colorante sólo en las Gram (-), mientras que en las Gram (+) el colorante queda retenido y las células permanecerán azules. Las células Gram (-) se teñirán después con un colorante de contraste (safranina) para que puedan observarse. Figura 11. Paredes celulares bacterianas. 31
- 32. Material Portaobjetos con frotis bacterianos previamente preparados a partir de cultivos aislados por cada equipo. Colorantes )a Solución de cristal violeta al 1 % )b Solución de safranina al 0.5 % )c Solución diluida de yodo (Lugol) )d Etanol-acetona (1:1) Microscopio y aceite de inmersión Papel filtro 1. Preparar frotis bacterianos. Prepare una extensión con cada una de las colonias aisladas en cultivo puro. 2. Teñir con cristal violeta (1 min). 3. Lavar con abundante agua el exceso de colorante. 4. Cubrir con lugol (1 min). 5. Lavar con agua el exceso de lugol. 6. Lavar la preparación con etanol-acetona hasta que observe que no arrastra más colorante de la muestra (10-20 segundos). 7. Lavar con agua para eliminar los restos de disolvente. 8. Teñir con safranina (1 min). 9. Lavar con agua para eliminar el colorante de contraste. 10. Secar la preparación entre dos hojas de papel filtro cuidando de no frotar, o bién secar por escurrimiento. 11. Examinar al microscopio con el objetivo de inmersión. 32
- 33. Figura 12. Tinción de Gram. RESULTADOS .1 Reportar la morfología de los microorganismos de cada una de las colonias observadas e identificar como grampositivas o gramnegativas. .2 Si cambiara el orden de empleo de los colorantes primario y de contraste, ¿cree que la tinción seguiría permitiendo distinguir entre grampositivas y gramnegativas? .3 ¿Cómo afectaría a las coloraciones finales si no se efectuara correctamente el lavado con los solventes orgánicos? .4 ¿Existe alguna relación entre el tipo de morfología y la tinción de Gram? PRACTICA 4. DETERMINACIÓN DE BACTERIAS COLIFORMES, COLIFORMES FECALES Y Escherichia coli POR LA TÉCNICA DE DILUCIONES EN TUBO MÚLTIPLE (NÚMERO MÁS PROBABLE O NMP) 33
- 34. OBJETIVO • Diferenciar los organismos coliformes totales de los microorganismos coliformes fecales. • Evaluar la calidad sanitaria de muestras de agua mediante la búsqueda de microorganismos coliformes totales, coliformes fecales y Escherichia coli. • Organizar e interpretar los resultados. GENERALIDADES Debido a que un gran número de enfermedades son transmitidas por vía fecal-oral utilizando como vehículo los alimentos y el agua, es necesario contar con microorganismos que funcionen como indicadores de contaminación fecal. Estos deben de ser constantes, abundantes, tener una sobrevivencia similar a la de los patógenos intestinales y deben de ser capaces de desarrollarse extraintestinalmente. El grupo coliforme es constante, abundante y casi exclusivo de la materia fecal, sin embargo, las características de sobrevivencia y la capacidad para multiplicarse fuera del intestino también se observan en aguas potables, por lo que el grupo coliforme se utiliza como indicador de contaminación fecal en agua; conforme mayor sea el número de coliformes en agua, mayor será la probabilidad de estar frente a una contaminación reciente. Los microorganismos coliformes constituyen un grupo heterogéneo con hábitat primordialmente intestinal para la mayoría de las especies que involucra. El grupo de bacterias coliformes totales comprende todos los bacilos Gramnegativos aerobios o anaerobios facultativos, no esporulados, que fermentan la lactosa con producción de gas en un lapso máximo de 48 h. a 35°C ± 1ºC. Este grupo está conformado por 4 géneros principalmente: Enterobacter, Escherichia, Citrobacter y Klebsiella. El grupo de coliformes fecales, está constituido por bacterias Gram- negativas capaces de fermentar la lactosa con producción de gas a las 48 h. de incubación a 44.5 ± 0.1°C. Este grupo no incluye una especie determinada, sin embargo la más prominente es Escherichia coli. La demostración y el recuento de organismos coliformes, puede realizarse mediante el empleo de medios de cultivo líquidos y sólidos con características selectivas y diferenciales. Escherichia coli es un bacilo corto Gram negativo que se encuentra clasificado dentro de la familia Enterobacteriaceae (bacterias entéricas), existe como comensal en el intestino delgado de humanos y animales. Sin embargo, hay algunas cepas de E. coli patógenas que provocan enfermedades diarreicas. Estas E. coli se clasifican con base en las características que presentan sus factores de virulencia únicos, cada grupo provoca la enfermedad por un mecanismo diferente. Las propiedades de adherencia a las células epiteliales de los intestinos grueso y delgado son codificadas por genes situados en plásmidos. De manera similar las toxinas son mediadas por plásmidos o fagos. Este grupo de bacterias se encuentra constituido por las siguientes cepas: E. coli enterotoxigénica (ETEC), E. coli enteropatógena (EPEC), E. coli enterohemorrágica (EHEC), E. coli enteroinvasiva (EIEC), E. coli enteroagregativa (EAEC) E. coli enteroadherente difusa (DAEC). Existen otras cepas que no han 34
- 35. sido perfectamente caracterizadas; de las cepas anteriores, las 4 primeras están implicadas en intoxicaciones causadas por el consumo de agua y alimentos contaminados. FUNDAMENTO La determinación de microorganismos coliformes totales por el método del Número más Probable (NMP), se fundamenta en la capacidad de este grupo microbiano de fermentar la lactosa con producción de ácido y gas al incubarlos a 35°C 1C durante 48 h., utilizando un medio de cultivo que contenga sales biliares. Esta determinación consta de dos fases, la fase presuntiva y la fase confirmativa. En la fase presuntiva el medio de cultivo que se utiliza es el caldo lauril sulfato de sodio el cual permite la recuperación de los microorganismos que se encuentren presentes en la muestra y que sean capaces de utilizar a la lactosa como fuente de carbono. Durante la fase confirmativa se emplea como medio de cultivo caldo lactosado bilis verde brillante el cual es selectivo y solo permite el desarrollo de aquellos microorganismos capaces de tolerar tanto las sales biliares como el verde brillante. La determinación del número más probable de microorganismos coliformes fecales se realiza a partir de los tubos positivos de la prueba presuntiva y se fundamenta en la capacidad de las bacterias para fermentar la lactosa y producir gas cuando son incubados a una temperatura de 44.5 0.1C por un periodo de 24 a 48 h. La búsqueda de Escherichia coli se realiza a partir de los tubos positivos de caldo EC, los cuales se siembran por agotamiento en medios selectivos y diferenciales (Agar Mac Conkey, Agar eosina azul de metileno) y posteriormente realizando las pruebas bioquímicas básicas (IMViC) a las colonias típicas. MEDIOS DE CULTIVO Y DILUYENTES 1. Para análisis de agua Para la preparación del medio de cultivo utilizado en la prueba presuntiva de muestras de agua o hielo, consultar el cuadro 1. • 5 ó 10 tubos de 22 x 175 mm con 10.0 mL de caldo lauril sulfato de sodio o caldo lactosado concentración doble o triple con campana de Durham. • 5 ó 10 tubos de 16 x 150 mm con 10.0 mL de caldo bilis verde brillante con campana de Durham. • 5 ó 10 tubos de 16 x 150 mm con 10.0 mL de caldo EC y campana de Durham. • Caldo EC MUG con campana de Durham. • 2 cajas Petri con agar para métodos estándar. • 2 cajas Petri con agar Eosina azul de metileno. • 6 tubos de 13 x 100 con 3.0 mL c/u de caldo RM-VPe • 3 tubos de 13 x 100 con 3.0 mL c/u de caldo triptona o agar SIM (opcional)e 35
- 36. • 3 tubos de 13 x 100 con 3.5 mL c/u de caldo citrato de Koser o citrato de Simmons (opcional) MATERIAL Y EQUIPO Mechero Perilla. Gradilla Balanza granataria. Pipetas de 10.0 mL estériles con tapón de algodón. Pipetas de 1.0 mL estériles con tapón de algodón. Pipetas Pasteur estériles Asa bacteriológica Portaobjetos Microscopio óptico Termómetro calibrado Baño de agua a 44.5° ± 0,1°C Incubadora a 35° ± 2,0ºC. Horno para esterilizar material de vidrio a 160-180°C Autoclave PROCEDIMIENTO 1. Agua 1.1 Prueba presuntiva • Agitar la muestra y transferir volúmenes de acuerdo con el cuadro 1, a cada uno de los tubos con caldo lauril sulfato de sodio que se hayan seleccionado. Agitar los tubos para homogeneizar la muestra. CUADRO 1. Preparación de inóculo con caldo lauril sulfato de sodio • In c u b a r los tubos a 35 ± 0,5°C. Examinar los tubos a las 24 h y observar si hay 36
- 37. formación de gas (desplazamiento del medio en la campana de Durham); si no se observa producción de gas, incubar 24 h más. 1.2 Prueba confirmativa de microorganismos coliformes totales • Transferir de 2 a 3 asadas de cada tubo positivo obtenido durante la prueba presuntiva, a otro tubo de 16 x150 mm que contiene caldo de bilis verde brillante, con campana de Durham. • Agitar los tubos para su homogeneización. • Incubar a 35 ± 2 oC durante 24 a 48 h • Registrar como positivos aquellos tubos en donde se observe turbidez (crecimiento) y producción de gas después de un período de incubación de 24 a 48 h. • Consultar la tabla 1 ó 2 de NMP para conocer el número más probable de organismos coliformes totales/100 mL. 1.3 Prueba confirmativa de microorganismos coliformes fecales. • Transferir de 2 a 3 asadas de cada tubo positivo obtenido durante la prueba presuntiva (caldo lauril sulfato de sodio) a un tubo de 16 x 150 mm, con caldo EC conteniendo campana de Durham. • Agitar los tubos para su homogeneización. • Incubar a 44.5 0.1C en incubadora o un baño de agua durante 24 a 48 h. • Registrar como positivos todos los tubos en donde se observe crecimiento y producción de gas después de un período de incubación de 24 a 48 h. • Consultar la tabla 1 ó 2 de NMP para conocer el número más probable de organismos coliformes fecales/ 100 mL. Control de calidad 1. Inocular a dos tubos con caldo EC una cepa de E. coli como control positivo y una de Enterobacter aerogenes como control negativo e incubar con las muestras. 1.4 Prueba confirmativa para Escherichia coli • Tomar una asada de cada uno de los tubos positivos en caldo EC y sembrar por estría cruzada en agar eosina azul de metileno para su aislamiento. • Incubar las placas invertidas a 35°C por 18-24 h. • Seleccionar dos colonias de cada placa con la siguiente morfología colonial: • Colonias con centro negro, planas con o sin brillo metálico. Si no hay colonias con morfología típica, probar una o más colonias lo más parecido E. coli de cada placa y sembrarlas en agar cuenta estándar para realizar las pruebas de morfología microscópica y pruebas bioquímicas. • Incubar las placas a 35°C por 18-24 h. • Hacer un frotis y teñirlo por Gram. Observar al microscopio la presencia de bacilos cortos o cocobacilos Gram-negativos. 37
- 38. CÁLCULOS Y EXPRESIÓN DE RESULTADOS 1. Calcular la densidad microbiana con base en el número más probable conforme al procedimiento señalado en la tabla 1, para estimar la población de bacterias coliformes totales, bacterias coliformes fecales y Escherichia coli de acuerdo con las diluciones empleadas. 2. Expresar en NMP/100 mL. En el caso de usar volúmenes de 20 mL de muestras de agua en 5 tubos o 10 mL de muestras de agua en 10 tubos, utilizar las siguientes tablas: TABLA 1. Índice del NMP con 95% de límite de confianza para varias combinaciones de resultados positivos y negativos cuando se usan 5 tubos con 20 mL de muestra de agua o hielo. 38
- 39. TABLA 2. Índice del NMP con 95% de límite de confianza para varias combinaciones de resultados positivos y negativos cuando se usan 10 tubos con 10 mL de muestra de agua o hielo. 39