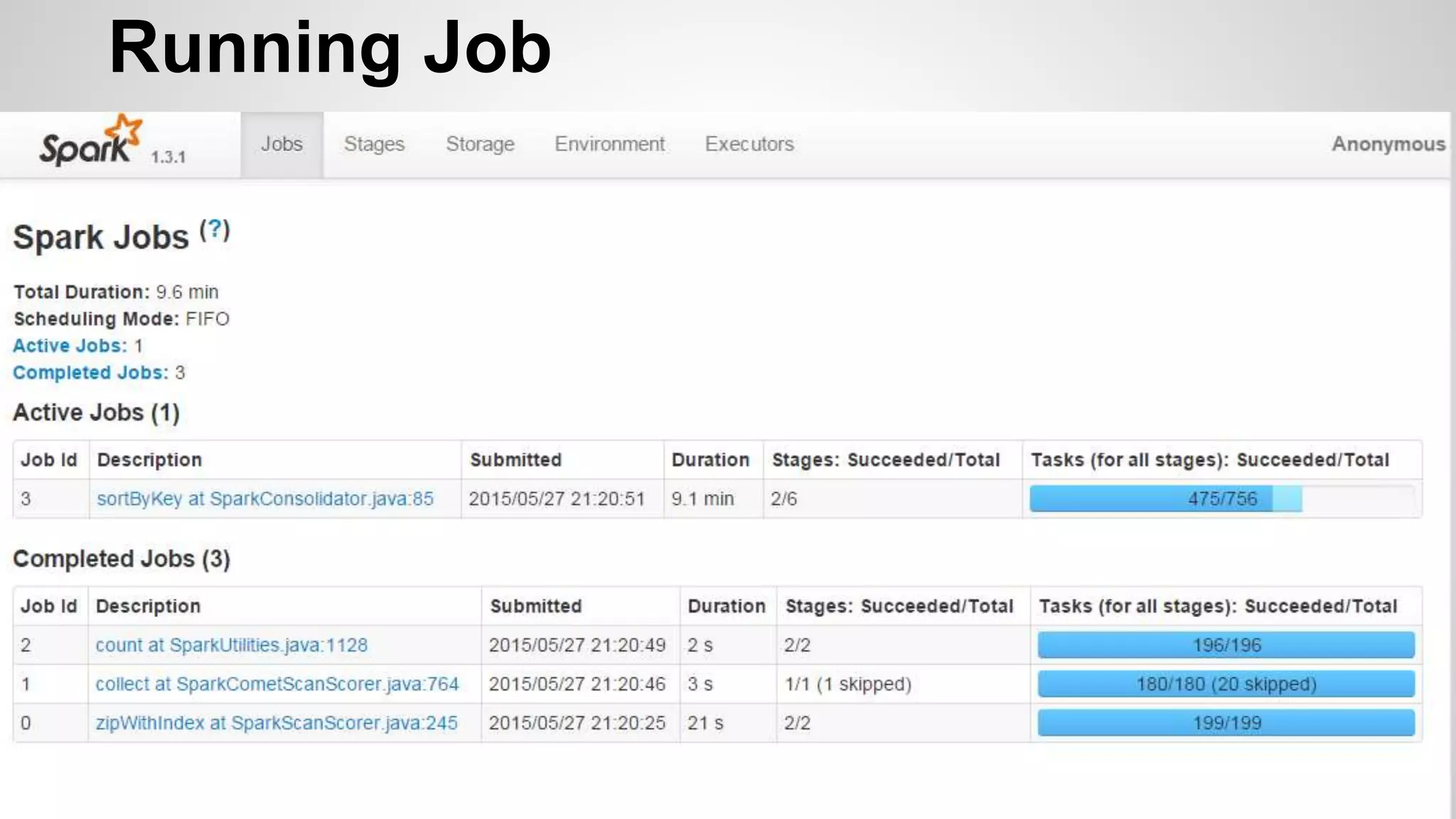
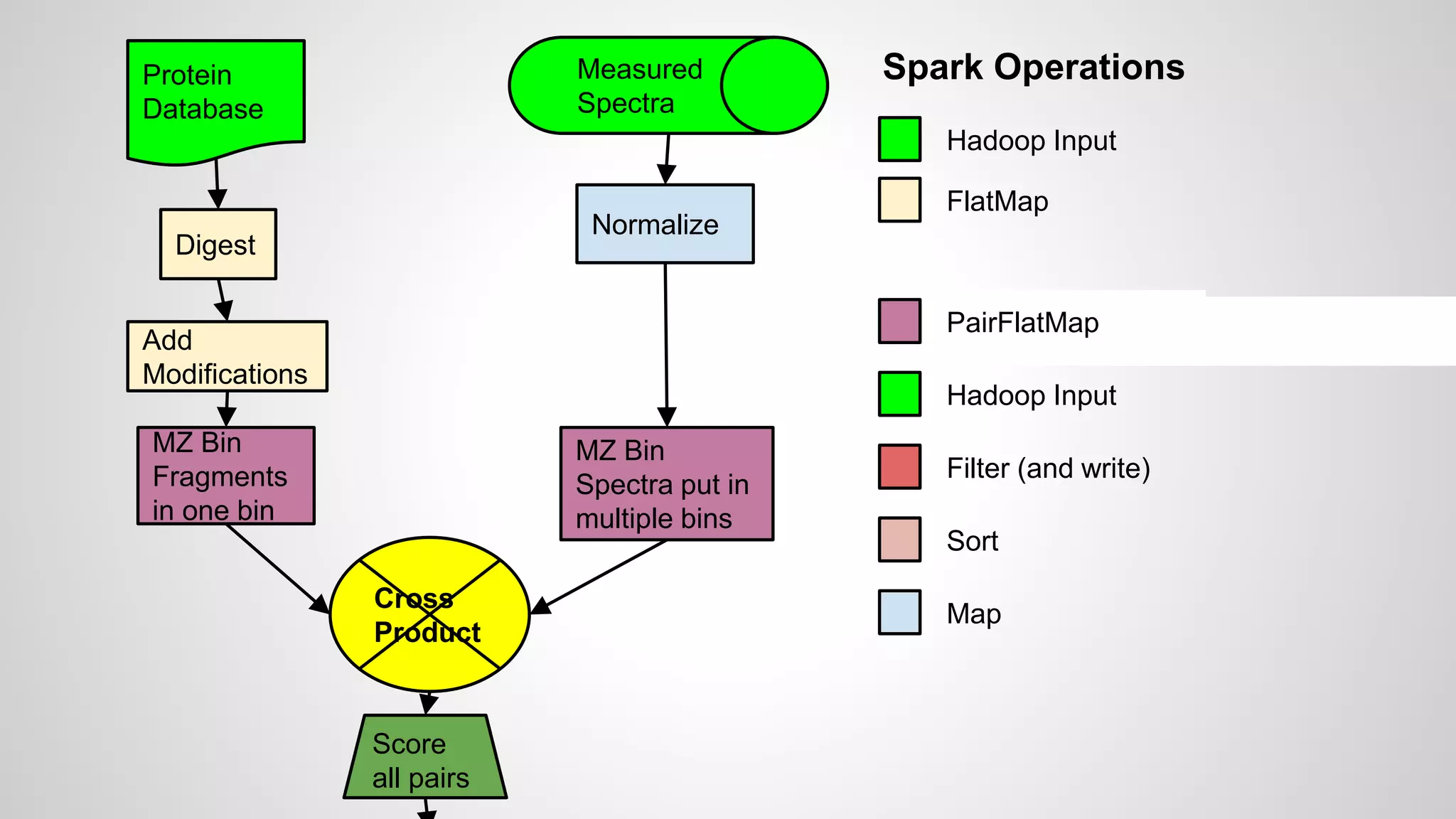
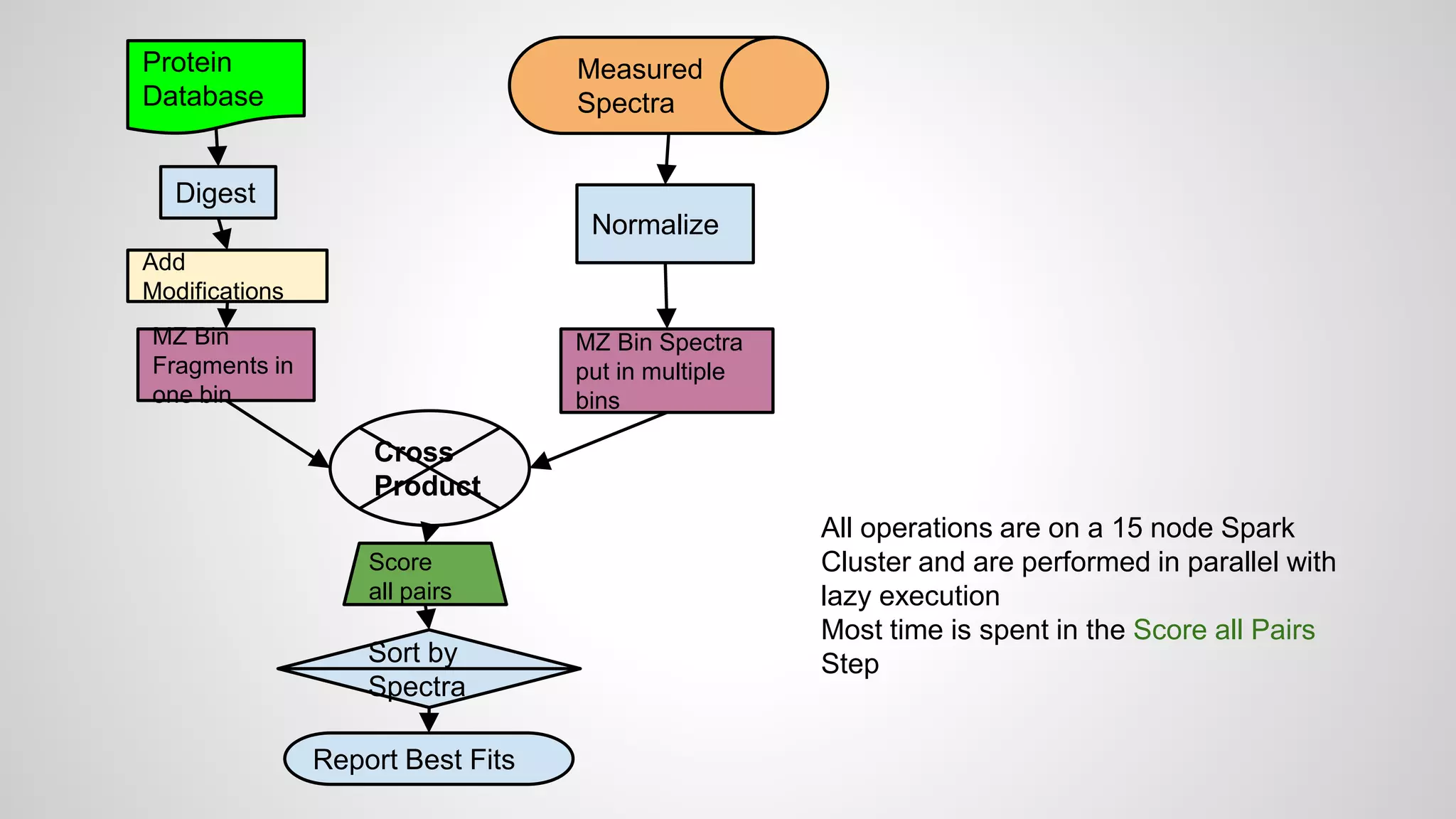
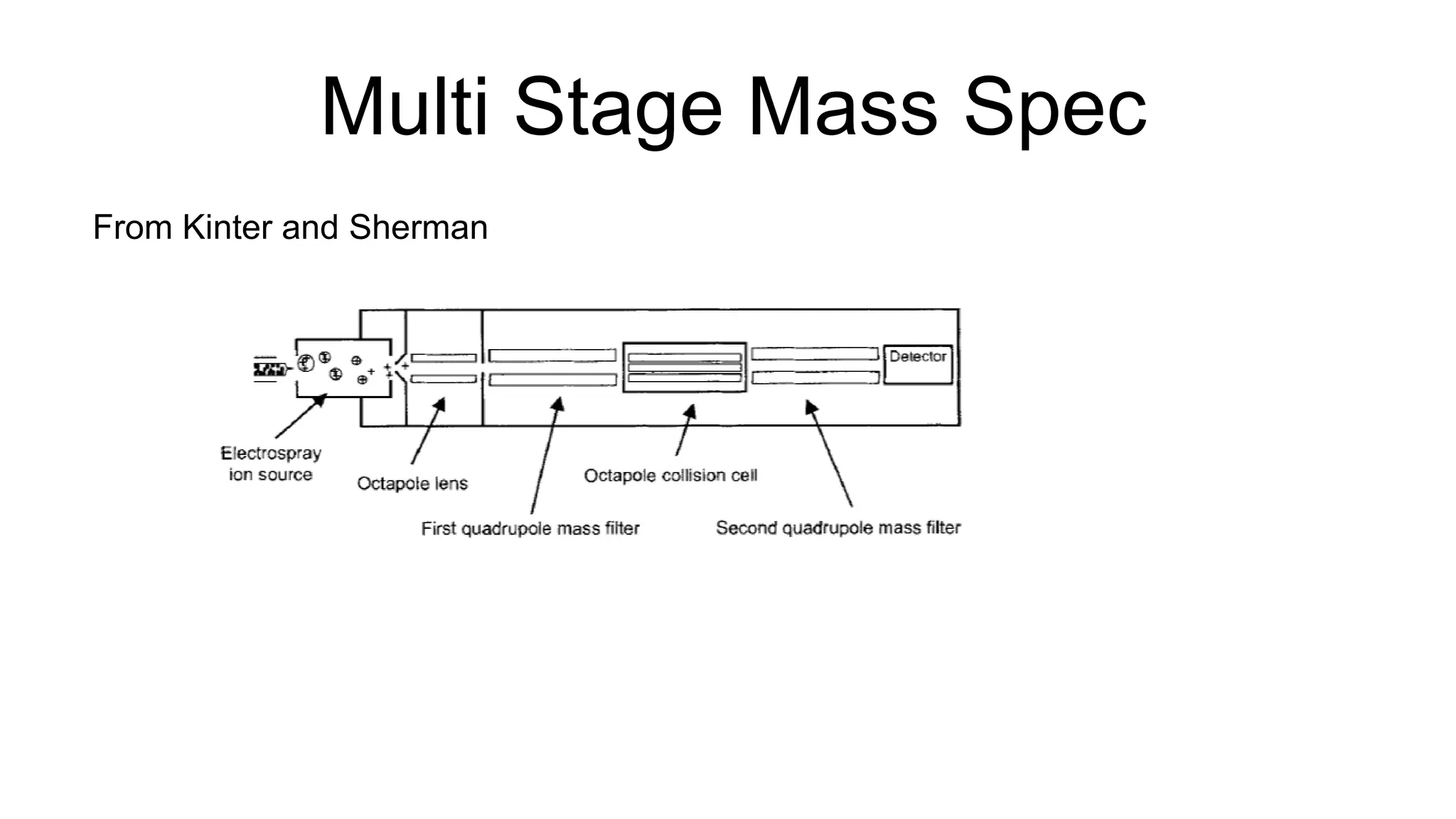
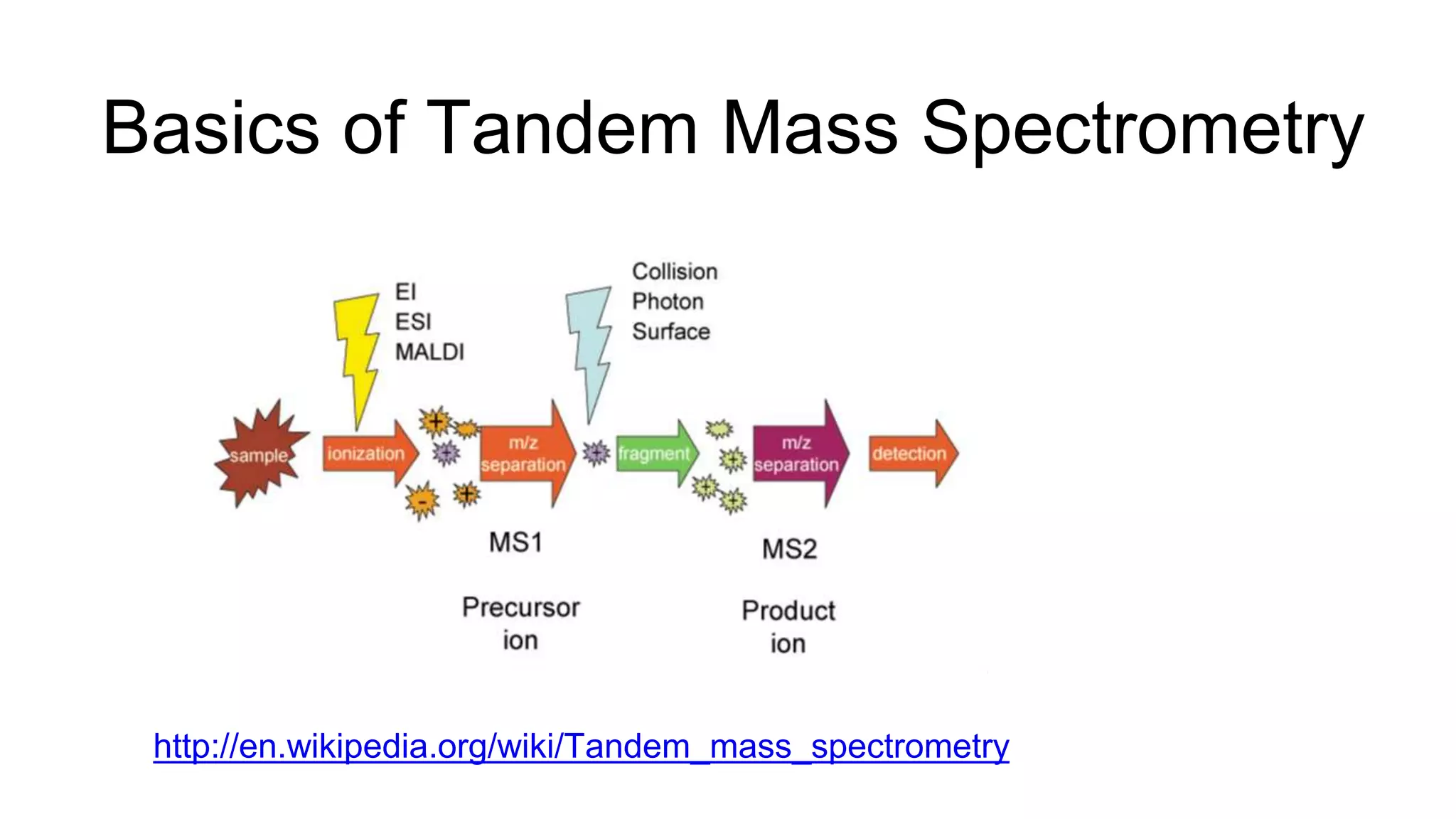
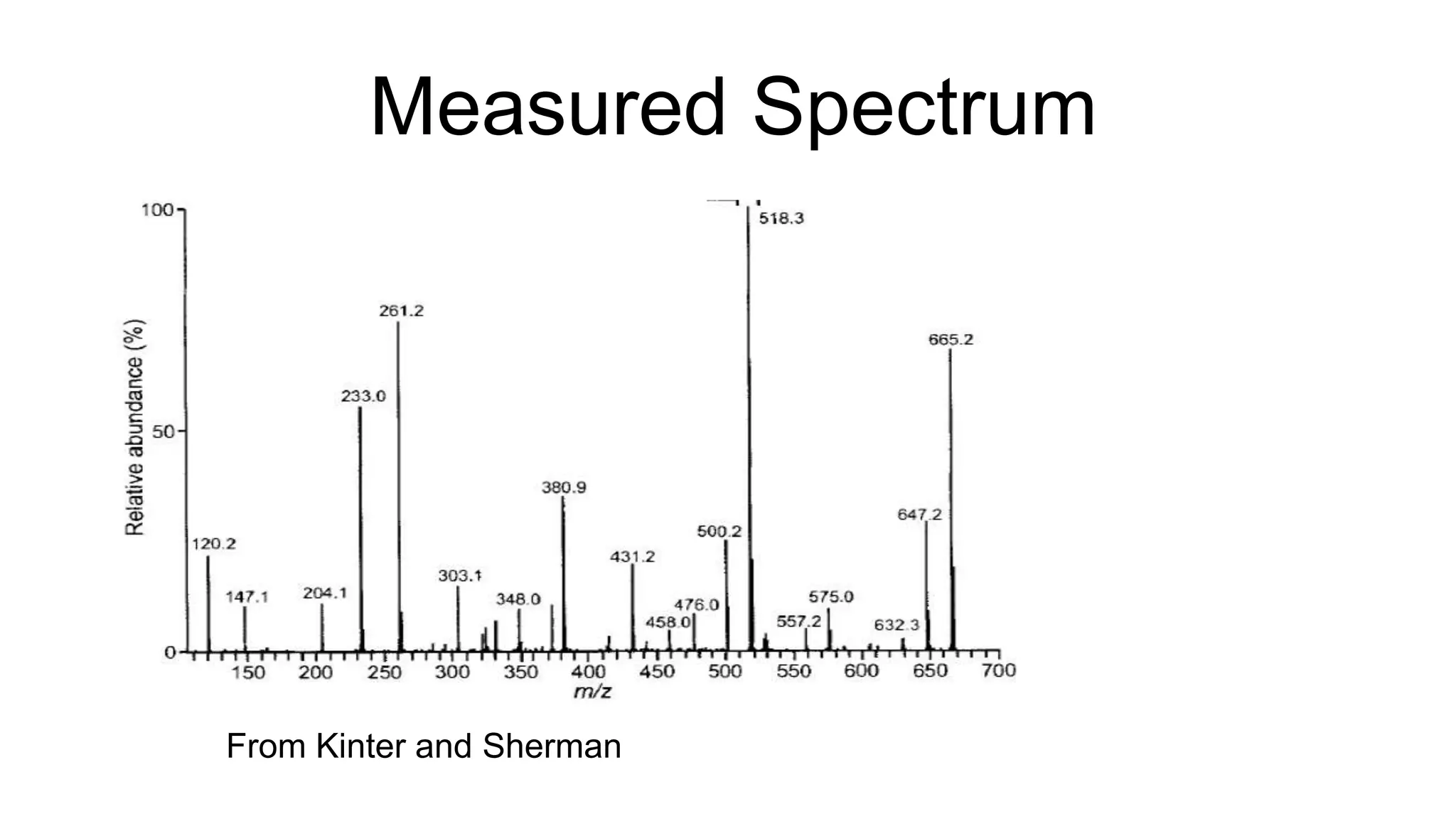
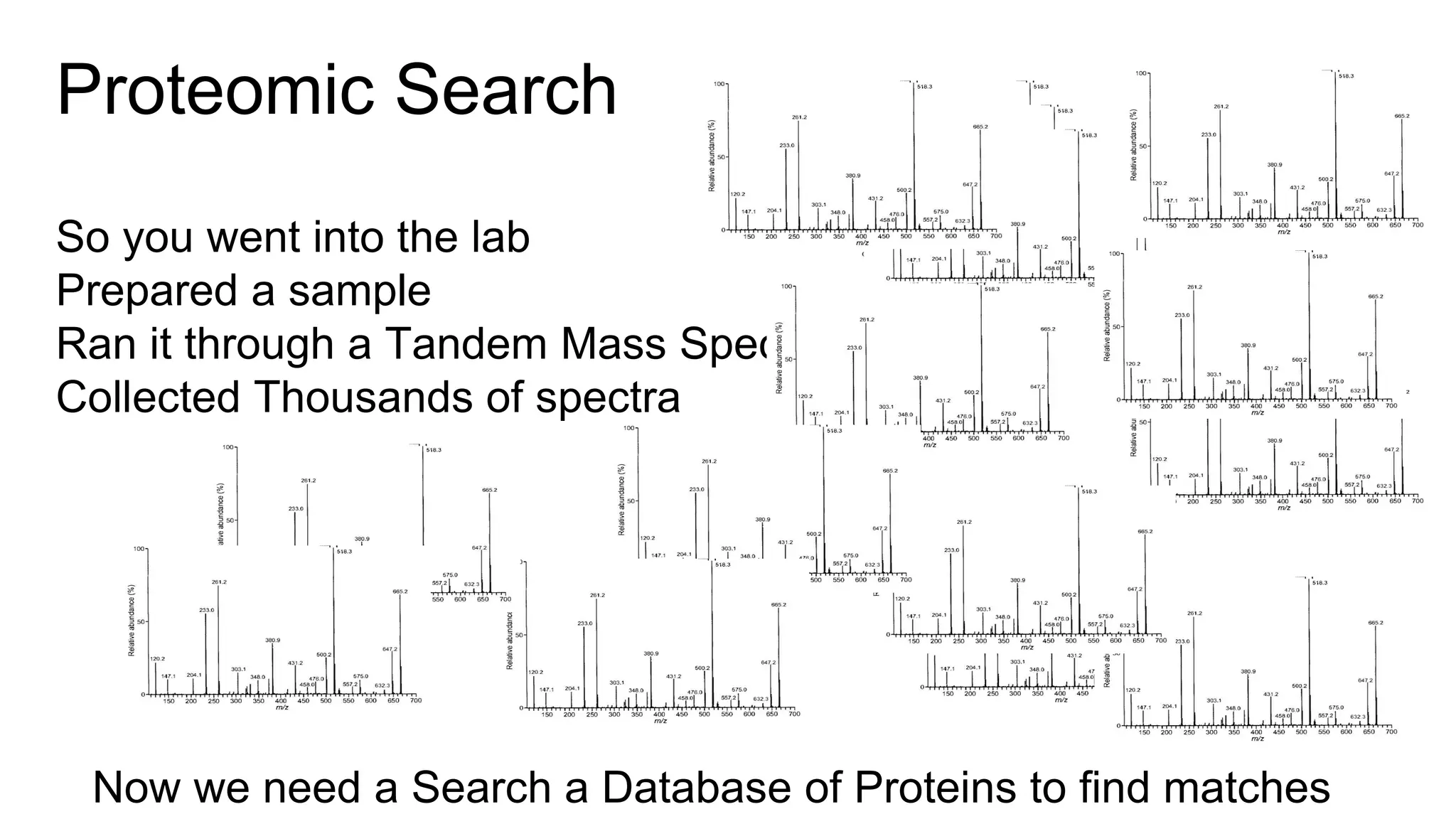
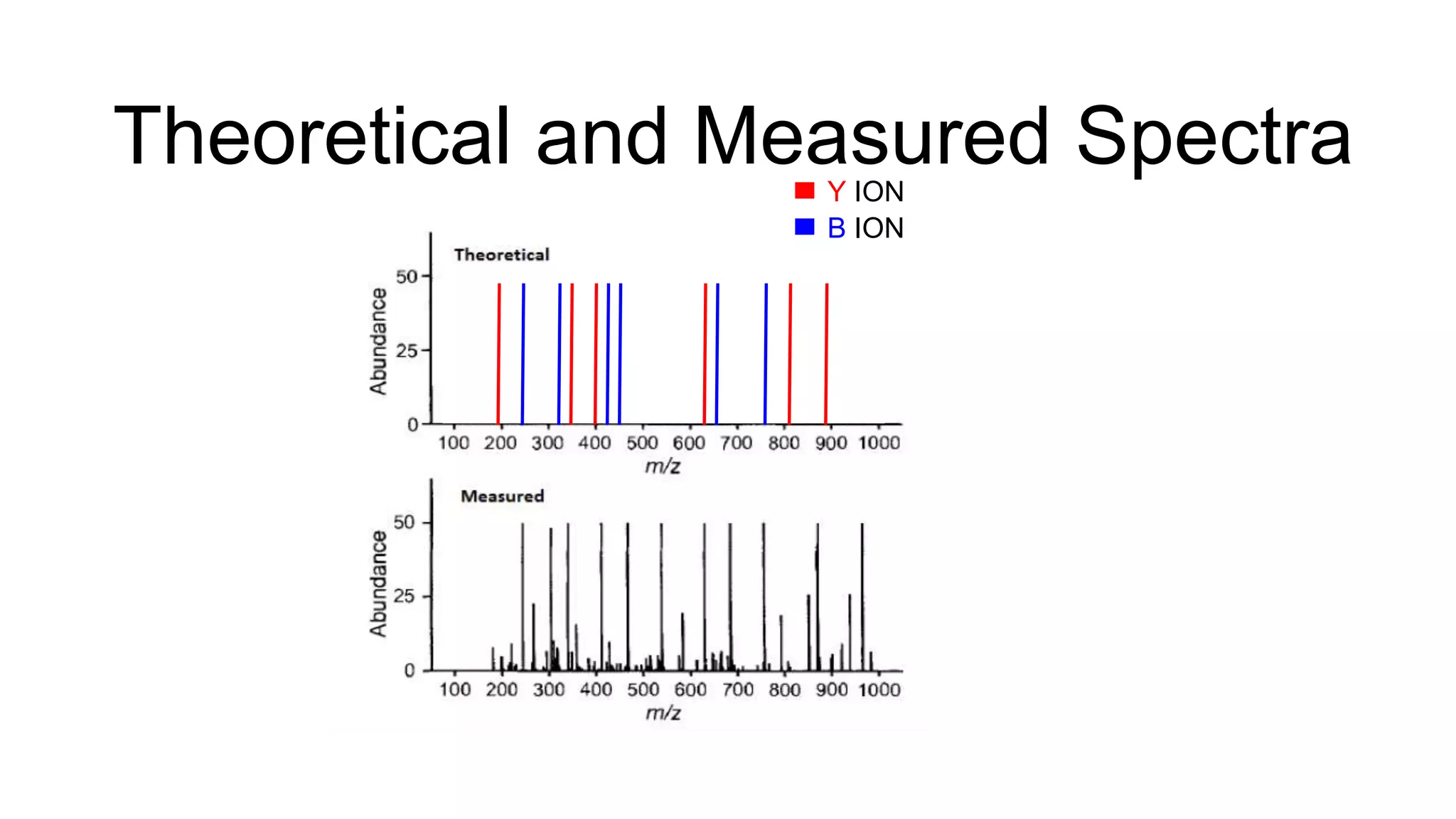
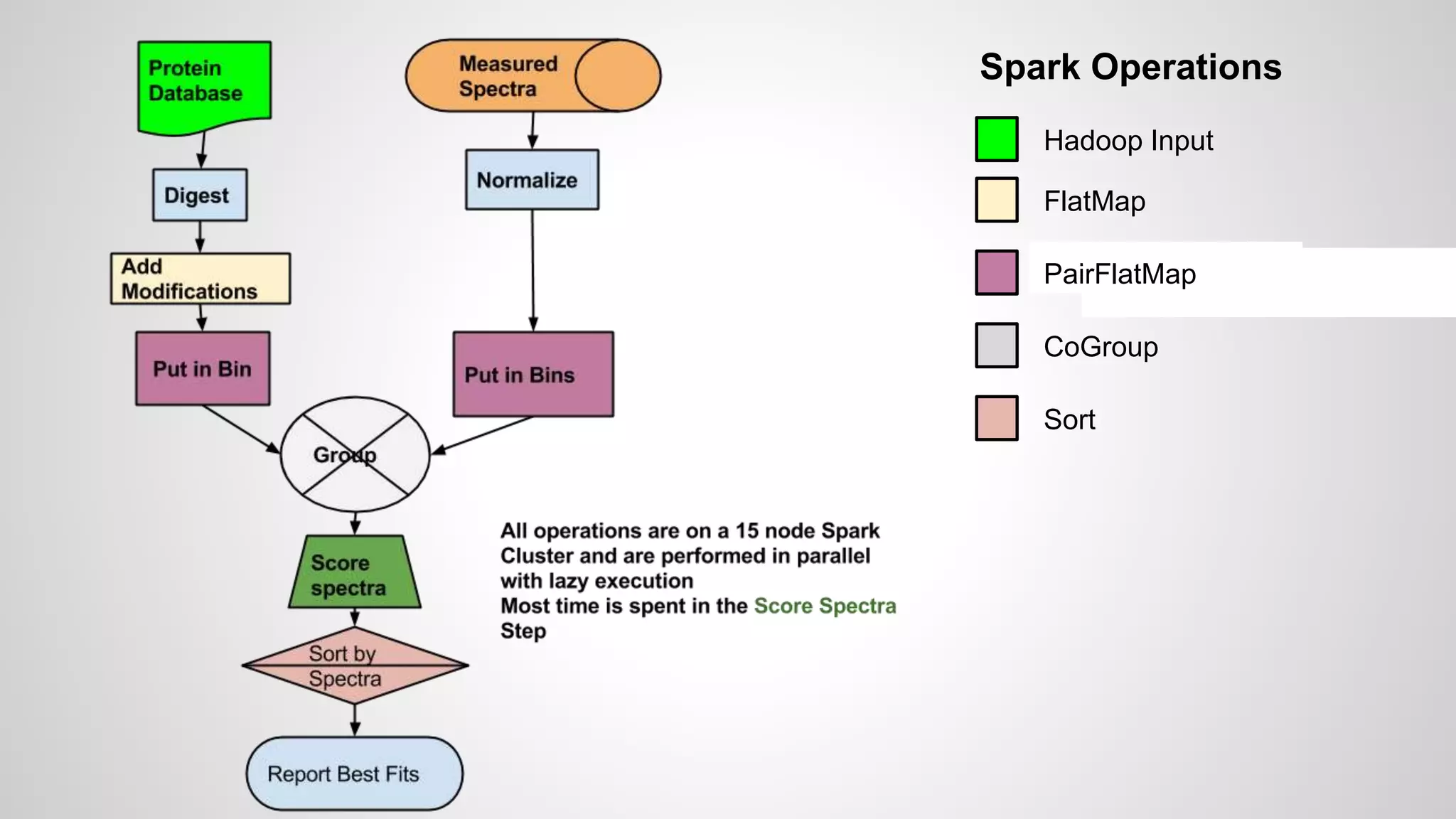
This document discusses using Apache Spark to parallelize proteomic scoring, which involves matching tandem mass spectra against a large database of peptides. The author developed a version of the Comet scoring algorithm and implemented it on a Spark cluster. This outperformed single machines by over 10x, allowing searches that took 8 hours to be done in under 30 minutes. Key considerations for running large jobs in parallel on Spark are discussed, such as input formatting, accumulator functions for debugging, and smart partitioning of data. The performance improvements allow searching larger databases and considering more modifications.









![Protein Database
● Starting with a database
● These are digested in silico to produce peptides
● Modifications may be added to produce a list of
peptides to search
● Every potential modification roughly doubles the search
space
IAM[15.995]S[79.966]GS[79.966]S[79.966]S
AIYVR
RGNTVLKDLK
IEFLNEAS[79.966]VMK
1360.63272
TVRAKQPSEK](https://image.slidesharecdn.com/useofsparkforproteomicscoring-seattlepresentation-150817163918-lva1-app6891/75/Use-of-spark-for-proteomic-scoring-seattle-presentation-13-2048.jpg)















![Sample Accumulator Use
JavaSparkContext ctx = new JavaSparkContext(sparkConf);
// make an accumulator
final Accumulator<Set<String>> wordsUsed = ctx.accumulator(new HashSet<String>(),
new SetAccumulableParam());
JavaRDD<String> lines = ctx.textFile(args[0]); // read lines
JavaRDD<String> words = lines.flatMap(new FlatMapFunction<String, String>() {
public Iterable<String> call(String s) {
List<String> stringList = Arrays.asList(s.split(" "));
wordsUsed.add(new HashSet<String>(stringList)); // accumulate words
return stringList;
}
});
… Finish word count](https://image.slidesharecdn.com/useofsparkforproteomicscoring-seattlepresentation-150817163918-lva1-app6891/75/Use-of-spark-for-proteomic-scoring-seattle-presentation-31-2048.jpg)