Download to read offline






















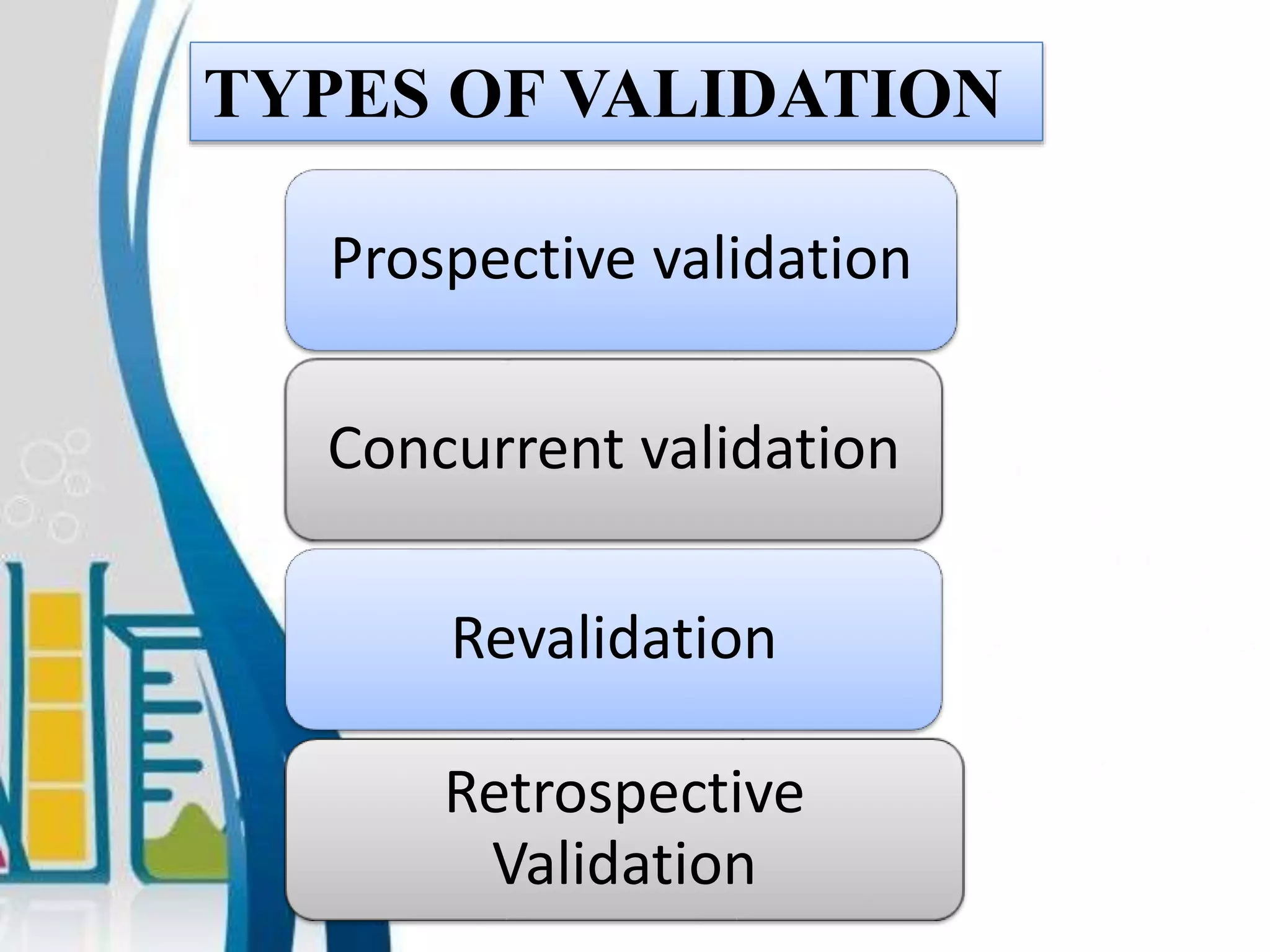











This document provides an overview of validation including: 1. The history and importance of validation in ensuring consistent quality production. Validation became a regulatory requirement after issues with sterility and product quality in the 1960s-1970s. 2. The scope of validation includes facilities, equipment, analytical methods, processes, cleaning, and personnel. 3. Key validation types are prospective, concurrent, retrospective, and revalidation which is required when changes are made. 4. A validation master plan outlines the entire validation program and assigns responsibilities. It provides a comprehensive guide for validation activities.










































































