This document defines bioavailability and bioequivalence and discusses related parameters and studies. It provides:
1) Definitions of bioavailability as the rate and extent to which the active drug is absorbed and available at the site of action, and bioequivalence as no significant difference in the rate and extent of absorption between test and reference drugs.
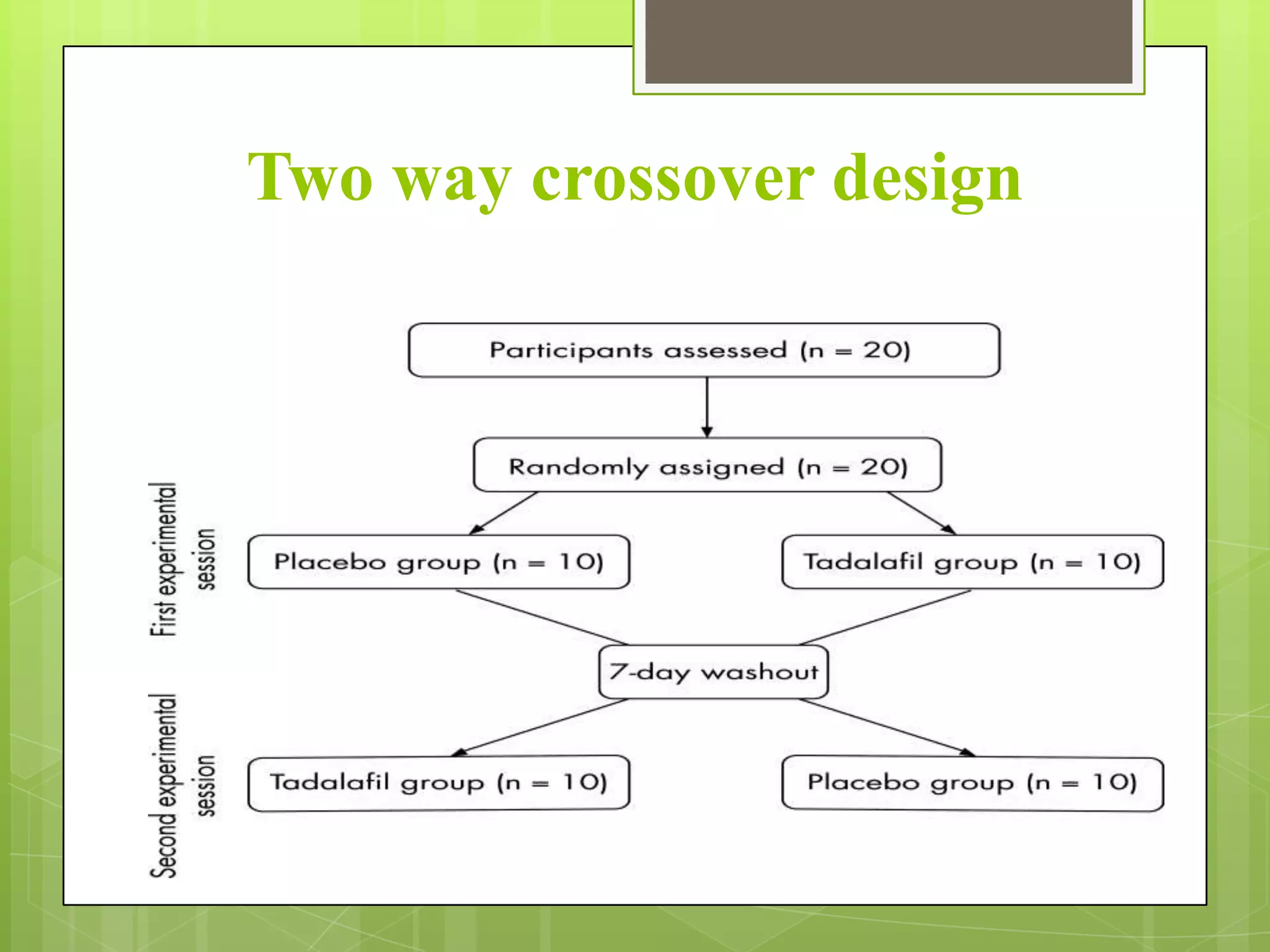
2) Reasons for conducting bioavailability/bioequivalence studies including changes to formulations, manufacturing processes, or for generic drug approval.
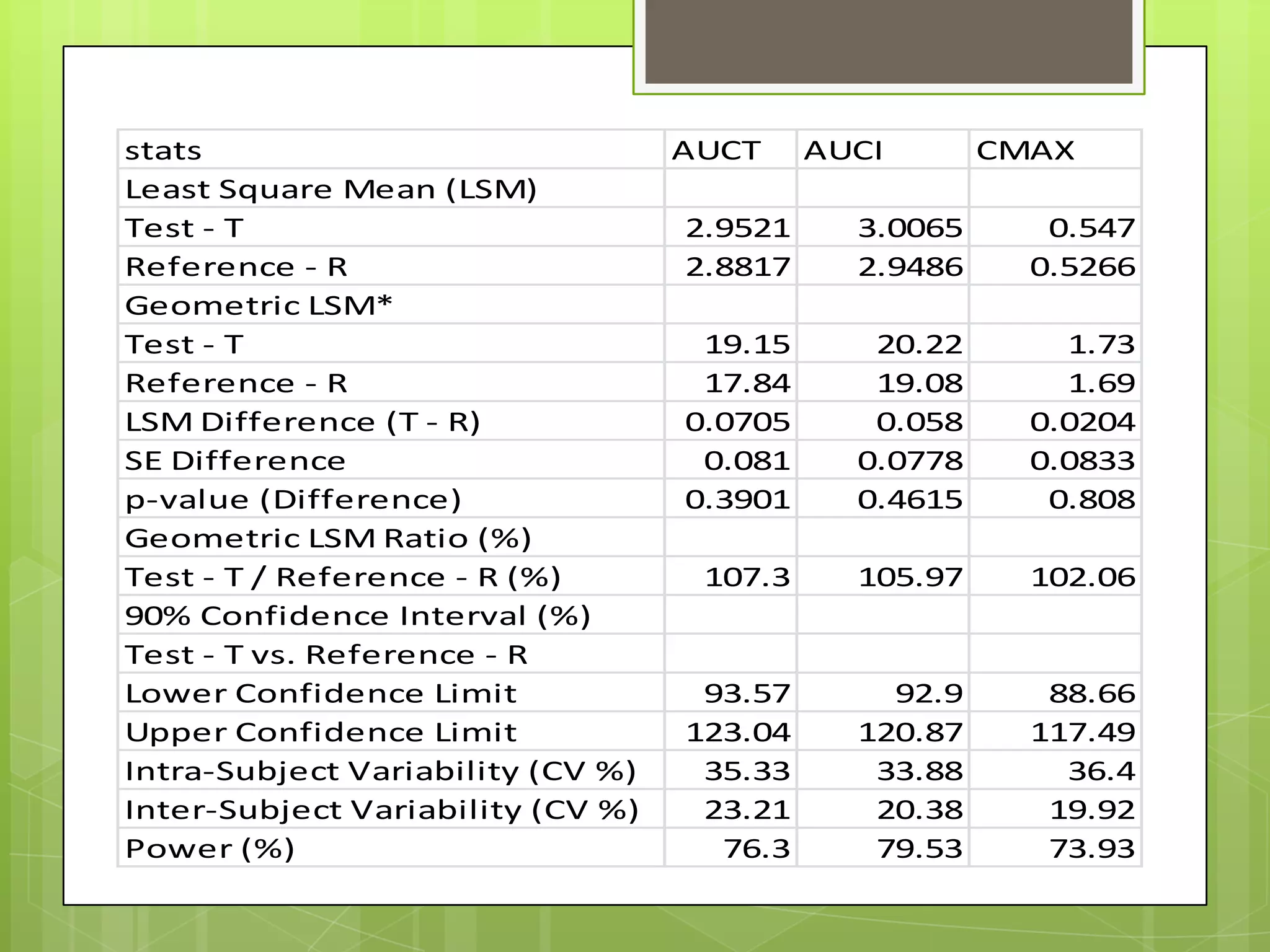
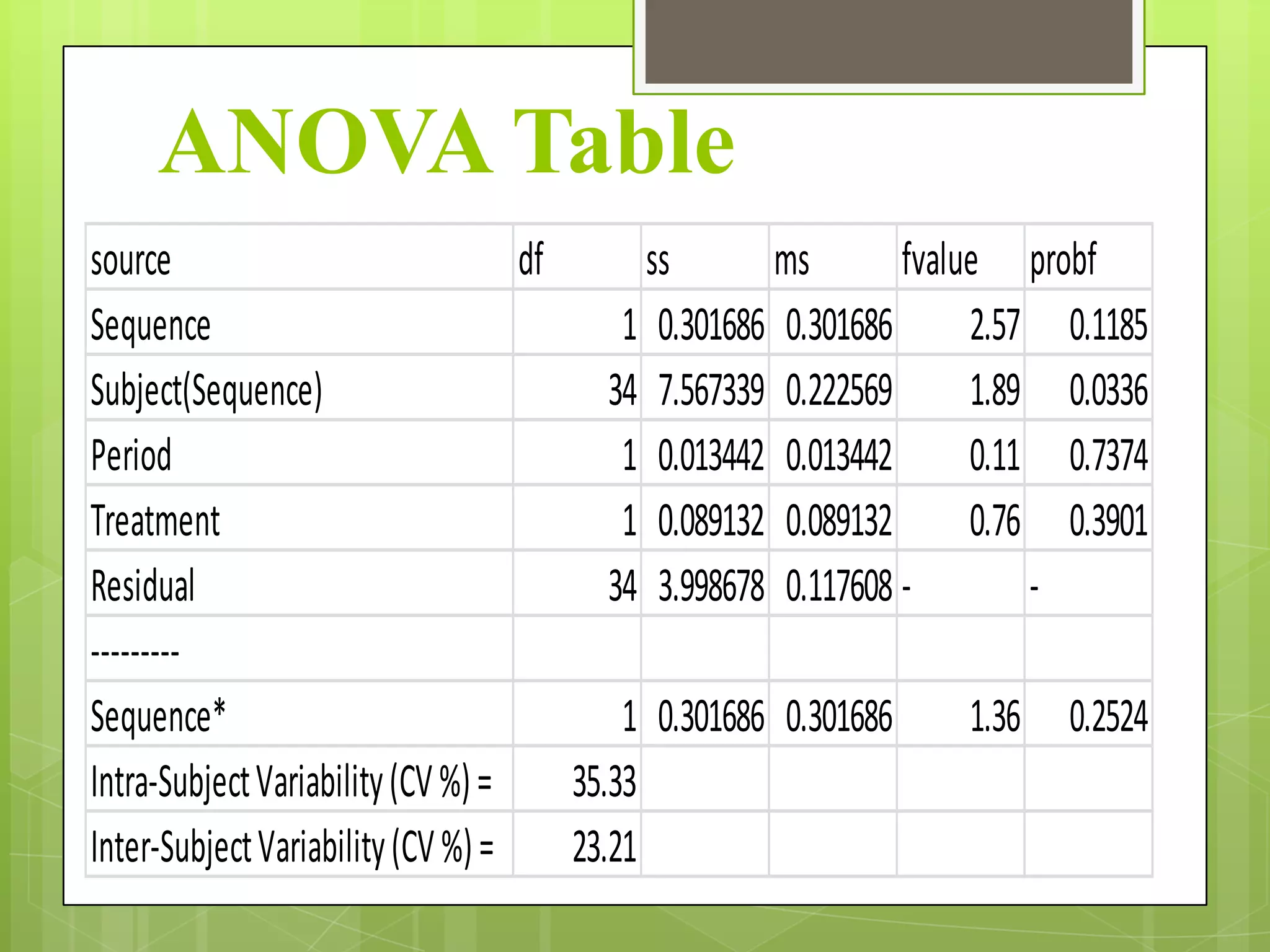
3) Key parameters analyzed in studies including Cmax, AUC, Tmax, and others to evaluate absorption rate and extent between test and reference drugs.




























![[Editage Seminar] Common language mistakes made by Japanese authors and essen...](https://cdn.slidesharecdn.com/ss_thumbnails/mbsj-v1-pdf-131202213136-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)









































![Rheumatic Fever CASE PRESENTATION [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationautosaved-251123182512-9d9b0da4-thumbnail.jpg?width=640&height=640&fit=bounds)



