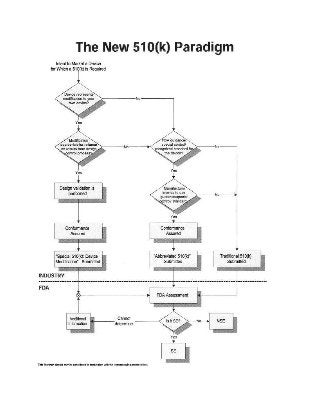
510k premarket submission is not required when someone is selling an incomplete device to another firm for further completion. If the device is manufactured outside the country and you are importing it, then you don't need 510 premarket submissions if the foreign manufacturer of that device has already submitted it. Read http://demandingbizservices.over-blog.com/2020/07/fda-510-k-submission-a-step-by-step-guide.html