Downloaded 85 times
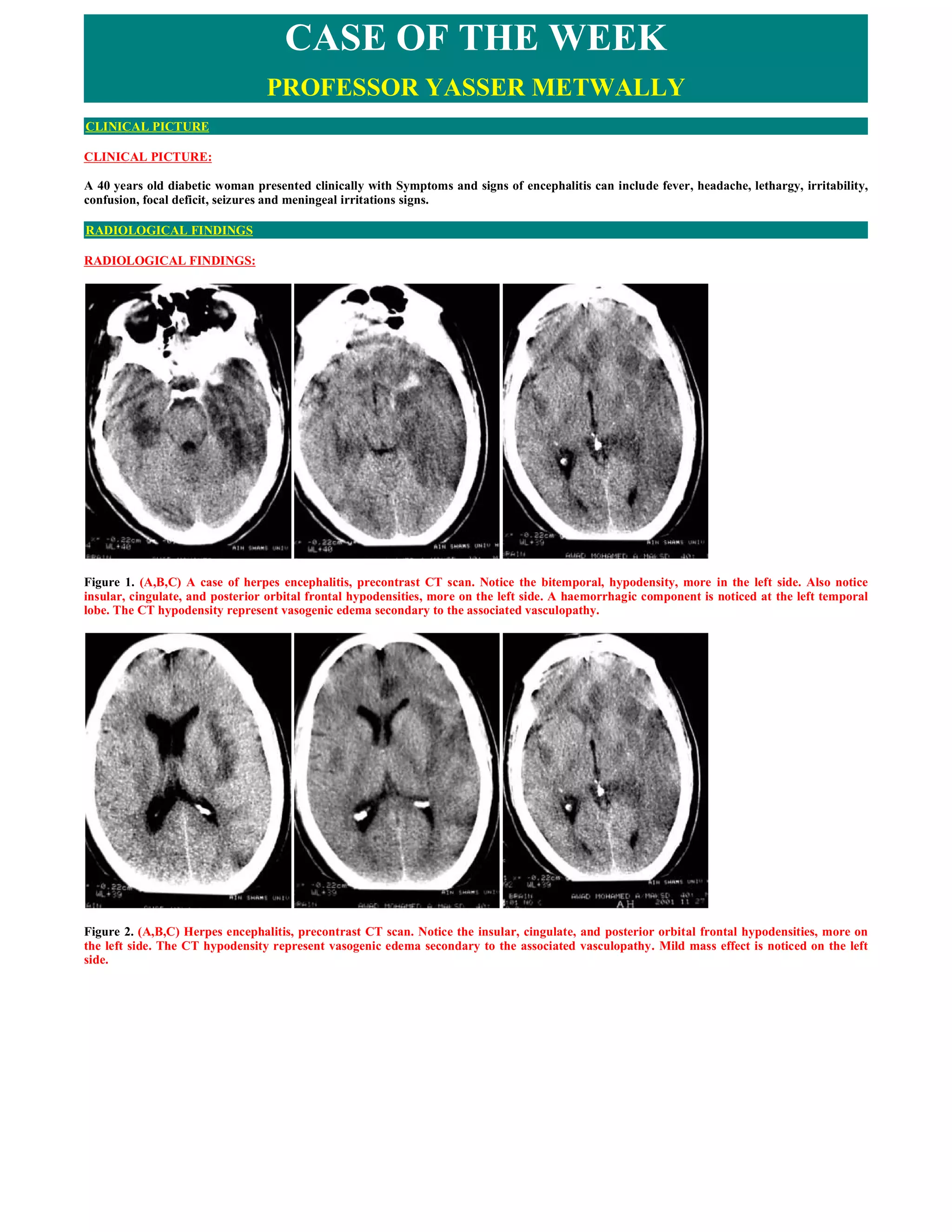
![Figure 3. (A,B,C) A case of herpes encephalitis, precontrast CT scan. Notice the bitemporal, hypodensity, more in the left side. Also notice
insular, cingulate, and posterior orbital frontal hypodensities, more on the left side. A haemorrhagic component is noticed at the left temporal
lobe. The CT hypodensity represent vasogenic edema secondary to the associated vasculopathy.
CSF examination
The opening pressure was elevated, and a CSF pleocytosis was observed. The patients had a mononuclear pleocytosis,(200 cells/mm3 (0.2 × 109
cells/l). CSF protein levels was elevated and the CSF glucose level was normal. Polymerase chain reaction (PCR) was positive for HSV-1 DNA .
Detection of HSV-1 DNA in CSF is sensitive and specific, and has become the diagnostic procedure of choice,[1]
Electroencephalography
Electroencephalography showed background disorganization with generalized and focal slowing, predominantly over the involved left temporal
region. Widespread, periodic and stereotyped sharp-wave and slow-wave complexes develop at intervals of 2–3 seconds was seen.[4]
DIAGNOSIS:
DIAGNOSIS: HERPES SIMPLEX TYPE 1 ENCEPHALITIS
DISCUSSION
DISCUSSION:
There are eight human herpesviruses (HHVs). Primary infection by any of the eight viruses, usually occurring in childhood, is either
asymptomatic or produces fever and rash of skin or mucous membranes; other organs might be involved on rare occasions. After primary
infection, the virus becomes latent in ganglia or lymphoid tissue. With the exception of HHV-8, which causes Kaposi's sarcoma in patients with
AIDS, reactivation of HHVs can produce one or more of the following complications: meningitis, encephalitis, myelitis, vasculopathy,
ganglioneuritis, retinal necrosis and optic neuritis. Disease can be monophasic, recurrent or chronic. Infection with each herpesvirus produces
distinctive clinical features and imaging abnormalities. This Review highlights the patterns of neurological symptoms and signs, along with the
typical imaging abnormalities, produced by each of the HHVs. Optimal virological studies of blood, cerebrospinal fluid and affected tissue for
confirmation of diagnosis are discussed; this is particularly important because some HHV infections of the nervous system can be treated
successfully with antiviral agents.
Herpesviruses are large double-stranded DNA viruses. Approximately 130 different herpesviruses have been identified, not only in mammals,
but also in frogs, lizards, birds, fish and mosquitoes. There are eight human herpesviruses (HHVs): herpes simplex virus (HSV-) 1, HSV-2,
varicella zoster virus (VZV or HHV-3), Epstein–Barr virus (EBV or HHV-4), cytomegalovirus (CMV or HHV-5), HHV-6, HHV-7, and HHV-8.
A characteristic feature of all herpesviruses is their ability to become latent, primarily in ganglia of the nervous system and lymphoid tissue. For
example, HSV and VZV become latent in neurons of ganglia, whereas EBV is latent in B lymphocytes. Most of the HHVs are neurotropic and
infrequently cause serious acute and chronic neurological disease of the PNS and CNS that might be monophasic, recurrent or chronic. Infection
with each herpesvirus produces different clinical features and imaging abnormalities, and many HHV infections can now be treated. This
Review focuses on the neurological complications of the HHVs, and discusses optimal virological tests to identify the etiological agent, along with
state-of-the-art treatments for these disorders.
Herpes Simplex Virus 1
Clinical Features
Encephalitis is the most serious neurological complication caused by HSV-1. Symptoms and signs of encephalitis can include fever, headache,
lethargy, irritability, confusion, focal deficit, aphasia and seizures, and they reflect virus replication with accompanying inflammation in the
medial temporal lobe and orbital surface of the frontal lobe. Progressive temporal lobe edema can lead to uncal herniation, with tachycardia,
hyperventilation, flexor (and later extensor) posturing, and a dilated pupil (usually on the side of the herniated temporal lobe). Survivors can be
left with a permanent seizure disorder, mental status changes, aphasia or motor deficit. Before the advent of aciclovir treatment, most patients
with HSV-1 encephalitis died.
Clinical investigations
Cerebrospinal fluid
The cerebrospinal fluid (CSF) is usually abnormal in patients with HSV-1 encephalitis. The opening pressure is often elevated, and a CSF
pleocytosis is observed in over 90% of patients, although its initial absence does not rule out HSV-1 encephalitis. Patients typically have a
mononuclear pleocytosis, with a mean of 200 cells/mm3 (0.2 × 109 cells/l). CSF protein levels are usually elevated, but CSF glucose levels are
usually normal. Increased CSF antibody to HSV-1, and a reduced serum:CSF antibody ratio, might help to diagnose HSV-1 encephalitis.
Antibody titers are not usually detected until 2 weeks or more after disease onset, however, and their practical value lies in retrospective](https://image.slidesharecdn.com/caseherp2-100117053949-phpapp02/75/Case-record-Herpes-simplex-type-1-encephalitis-2-2048.jpg)
![presumptive diagnosis. HSV-1 can be isolated from cerebral biopsy or autopsy material, but isolation of the virus from CSF is rare. Polymerase
chain reaction (PCR) detection of HSV-1 DNA in CSF is sensitive and specific, and has become the diagnostic procedure of choice,[1] although
PCR results can be negative during the early stages of disease.[2] Pathological changes in HSV-1 encephalitis include localized inflammation,
hemorrhagic necrosis and formation of inclusion bodies.
Herpes Simplex Virus 1 and Bell's Palsy
Virologic analysis of endoneurial fluid obtained during decompression surgery revealed HSV-1 DNA in 11 out of 14 patients with Bell's palsy.
[23] As seropositivity to HSV-1 is well established by adult life, when Bell's palsy is most common, the palsy probably reflects virus reactivation
from latency in the geniculate ganglion[24] rather than primary infection. The mechanism through which the virus damages the facial nerve is
unknown.
Electroencephalography
Electroencephalography in patients with HSV-1 encephalitis shows background disorganization with generalized or focal slowing,
predominantly over the involved temporal region.[3] In some patients, widespread, periodic and stereotyped sharp-wave and slow-wave
complexes develop at intervals of 2–3 seconds.[4] Bilateral periodic complexes appear if both hemispheres are involved and, although they are
seen in other CNS disorders, the presence of such complexes in the setting of fever and rapidly progressive neurological disease is strongly
indicative of HSV-1 encephalitis.
Neuroimaging
CT scanning reveals hypodense lesions involving the medial temporal regions, with a sharp transition from the hypodense temporal lesion to
normal basal ganglia.[5] Edema and mass effect occur in 80% of cases, and lesions enhance with contrast. MRI reveals a decrease in T1 signal
and an increase in T2 signal in the orbitofrontal and medial temporal lobes and insulae, with abnormalities appearing earlier and more
frequently than with CT.[6] With modern neuroimaging techniques, HSV-1 encephalitis is only rarely confused with cerebritis, abscess, tumor
or infarction. Figure 1 shows typical CT and MRI changes in patients with HSV-1 encephalitis compared with imaging abnormalities in VZV
vasculopathy (see below) and other viral encephalitides.
Figure 1. Typical changes seen on CT and MRI in patients with herpesvirus infections. (A,B,C) Herpes simplex virus 1 (HSV-1) encephalitis: T2-
weighted MRI brain scan demonstrates bilateral involvement of temporal lobes. The exaggerated signal does not extend beyond the insular
cortex (thin arrow), but does involve the cingulate gyrus (thick arrow).
Figure 2. MRI study in a patient suffering from herpes encephalitis, A,B,C MRI T1 precontrast, D,E MRI T2, F,G,H,I are FLAIR MRI images.](https://image.slidesharecdn.com/caseherp2-100117053949-phpapp02/75/Case-record-Herpes-simplex-type-1-encephalitis-3-2048.jpg)
![Notice the T1 hypointensity predominantly involving the bitemporal cortex more on the right side, the insular, the occipital orbital frontal (F)
and the cingulate cortex, again more on the right side. The T1 involved zones are hyperintense on the T2 and FLAIR studies. Notice that the
encephalitic process is predominately cortical (A,B,C). Temporal lobes involvement is more medially than laterally. The MRI signal changes are
predominantly due to vasogenic edema, the signal intensity of edema is different from the CSF in flair studies.
Pathogenesis and Latency
Primary HSV-1 infection often results in painful skin or mucosal lesions, but can also be asymptomatic. Virus replicates at the portal of entry,
usually oral or genital mucosal tissue, leading to infection of sensory nerve endings. Virus is then transported to regional ganglia where it
establishes latency. Recurrence is directly related to the severity of primary infection, as reflected by the size, number and spread of lesions. The
mechanism by which HSV-1 infects the CNS to cause encephalitis has not been definitively established. The predilection of HSV-1 for the orbital
surface of the frontal lobe and medial surface of the temporal lobe indicates that virus might spread from olfactory mucosa through the
cribiform plate of the ethmoid bone into the anterior fossa. Latent virus in the trigeminal ganglia might also reactivate and spread via tentorial
nerves that innervate the meninges of the anterior and middle cranial fossa.
Figure 3. A, This is a gross photograph of a section of brain showing multiple small, punctate hemorrhages throughout the brain parenchyma,
especially the insula and the medial temporal cortex, in a case of herpes encephalitis. B, A case of herpes encephalitis with gross haemorrhagic
necrosis of the temporal lobe.
HSV-1 latency is restricted to cranial nerve ganglia, as indicated by spontaneous, recurrent outbreaks of vesicles on the mouth, or by isolation of
HSV-1 from postmortem explants of human trigeminal,[7,8] nodose, vagal[9] and ciliary[10] ganglia. HSV-1 DNA sequences have been detected
in human thoracic ganglia[11] and brain,[12] but virus has never been recovered from these sites. Most humans harbor latent HSV-1.[11]
Neurons are the exclusive site of HSV-1 latency in human ganglia, and also in mouse and rabbit models. Studies of latently infected mouse
ganglia have revealed HSV-1 DNA in 1–30% of neurons.[13] In humans, the latent HSV-1 DNA copy number ranges from less than 300
copies/105 cells to over 10,000 copies/105 cells in trigeminal ganglia,[14] and has a mean of 176,705 ± 255,916 copies/105 cells in vestibular
ganglia, 9,948 ± 22,066 copies/105 cells in geniculate ganglia, and 3,527 ± 9,360 copies/105 cells in cochlear ganglia.[15] During latency, HSV-1
DNA is endless—that is, circular or concatameric[16]—and does not integrate into the host genome.
Figure 4. Right, Coronal section showing mesial temporal lobe, insular necrosis in a case of herpes simplex virus (HSV) encephalitis. Left,
Typical homogenous, eosinophilic Cowdry A inclusion in HSV encephalitis.
In ganglia latently infected with HSV-1, only one region of the viral genome is abundantly transcribed. This region encodes the latency-
associated transcripts (LATs). The LAT domain is transcriptionally complex, and although the predominant species that accumulates in the
neuronal nucleus during latency is a 2.0 kb stable intron,[17] other RNA species are also transcribed, including a number of lytic or acute-phase
transcripts. No LAT protein has ever been detected. LAT-deficient HSV-1 mutants become latent,[18] and splicing of the primary LAT
transcript appears to be important in promoting latency.[19] The function of LATs is unknown, but they might protect neurons from apoptosis,
[20] perhaps by acting through microsomal RNA.[21]](https://image.slidesharecdn.com/caseherp2-100117053949-phpapp02/75/Case-record-Herpes-simplex-type-1-encephalitis-4-2048.jpg)
![Figure 5. A precontrast CT scan showing a case of herpes encephalitis, notice the left medial temporal hemorrhagic zone surrounded by edema.
Figure 6. Section at the level of the anterior commissure showing softening and disruption
of the right lateral temporal lobe with some hemorrhage in a case of herpes encephalitis
HSV-1 reactivates after ultraviolet (UV) light stimulation, epinephrine iontophoresis, hyperthermia, or even social stress. Variables include
duration of stimuli and latent virus DNA burden. Accumulating evidence from rodent models indicates that HSV-1 infection suppresses the
hypothalamic–pituitary–adrenal axis during primary infection, and that this stress-induced suppression has a role in virus reactivation. The
relevance of animal models to humans must, however, always be questioned. For example, guinea pigs vaccinated with HSV-2 glycoprotein D
were protected from reactivation, whereas subunit vaccines were only marginally effective in human trials.[22]
Figure 7. Schematic of the typical areas of involvement by herpes simplex virus.
Note the propensity of involvement in the medial temporal lobes, in the insular
cortex and the cingulate cortex.
Treatment
Intravenous aciclovir (10 mg/kg body weight three times a day for 14–21 days) is the standard treatment for HSV-1 encephalitis in adults.
Steroids are sometimes given as well, although data regarding the efficacy of this approach remain anecdotal. Cognitive impairment and seizures
are significant neurological sequelae in treated patients.
Acyclovir, an acyclic analogue of guanosine, is the antiviral drug of choice for treatment of neurologic disease associated with herpes simplex
virus-1 or herpes simplex virus-2 infection. Its mechanism of action depends on a virus-specified thymidine kinase that phosphorylates acyclovir
to its monophosphate derivative. Acyclovir monophosphate is phosphorylated further by cellular kinases to acyclovir triphosphate, which binds
to virus- induced DNA polymerase, acting as a DNA chain terminator. Because acyclovir is taken up selectively by virus-infected cells, the
concentration of acyclovir triphosphate is 40 to 100 times higher in infected cells than in uninfected cells. In addition, virus-induced DNA
polymerase exhibits a 10- to 30-fold greater affinity for acyclovir triphosphate than do cellular polymerases. For these reasons, the drug exhibits
low toxicity for uninfected cells and is therefore well tolerated clinically. Acyclovir is highly specific for the herpes virus and a clinical response
to acyclovir has also a diagnostic implications as its action depends on a virus-specified thymidine kinase. No clinical response will occur if the
offending organism is other than the herpes virus.
The relative safety of acyclovir has led to the common practice of presumptive treatment of patients who exhibit typical clinical signs and
symptoms of the disease with typical radiological findings. The outcome of acyclovir therapy depends on the age of the patient and level of
consciousness when treatment is initiated.](https://image.slidesharecdn.com/caseherp2-100117053949-phpapp02/75/Case-record-Herpes-simplex-type-1-encephalitis-5-2048.jpg)
![Massive brain edema is observed in most of patients. Corticosteroids are probably not useful in the treatment of edema associated with HSE.
Data from animal studies have shown even potentiation of disease. 48 Brain edema in all patients responded dramatically, even when massive, to
acyclovir It seems quite logic that treatment of the cause of brain edema is the best treatment of brain edema.
The goals of using antivirals are to shorten the clinical course, prevent complications, prevent development of latency and subsequent
recurrences, decrease transmission, and eliminate established latency.
Table 1. Acyclovir therapy
Acyclovir (Zovirax)- Has demonstrated inhibitory activity against both HSV-1
and HSV-2 and is taken up selectively by infected cells; rate of mortality from
Drug Name
HSE before use of acyclovir was 60-70%—since acyclovir, it is approximately
30%.
Adult Dose 10 mg/kg/dose IV or 500 mg/m2/dose IV q8h
Pediatric Dose Administer as in adults
Contraindications Documented hypersensitivity
Half-life prolonged and toxicity increased by concomitant probenecid or
Interactions
zidovudine
Pregnancy B - Usually safe but benefits must outweigh the risks.
Use caution in patients with renal failure or receiving other nephrotoxic drugs
Precautions
concurrently
Figure 8. A case of herpes encephalitis treated with acyclovir, notice the massive brain oedema before treatment (A), also notice the gradual
reduction of brain oedema in (B) and complete resolution of brain oedema in (C) following treatment with acyclovir, this was coupled with
dramatic clinical improvement. Notice the residual frontal infarction that became apparent after resolution of the encephalitic process.
Table 2. Clinical Features of Virus Reactivation
Feature Herpes simplex Varicella zoster
Reactivation Multiple times Once
age Young Elderly
Rash Localized Dermatomal
Aetiology Sunlight, trauma Immunosuppression
Herpes Simplex Virus 2
Clinical Features
HSV-2 causes genital herpes, and neurological complications include aseptic meningitis and recurrent radiculopathy. HSV-2 also causes myelitis
in immunocompromised individuals. Typical features of HSV-2 meningitis are headache, fever, stiff neck, and lymphocytic pleocytosis in the
CSF. HSV-2 meningitis might be preceded by pelvic inflammatory disease, genital pain or both, and clinical examination should include a search
for vesicular lesions over the external genitalia, and also for lesions in the vagina or on the cervix. It is important to recognize, however, that
many patients with HSV-2 meningitis do not have active genital lesions at the time of meningitis, and often have no known history of genital
herpes. HSV-2 was shown to cause aseptic meningitis in one patient with recurrent dermatomal distribution skin lesions.[25] PCR has shown
that HSV-2,[26] and less often HSV-1,[27] causes benign recurrent lymphocytic meningitis, which is usually self-limited.
HSV-2 encephalitis occurs most often in newborn babies and, more rarely, in immunocompromised adults. Approximately 5% of HSV
encephalitis cases in immunocompetent adults are also attributable to HSV-2. In infants and immunocompromised adults, HSV-2 encephalitis is
diffuse, unlike the focal lesions produced by HSV-1 infection. Nevertheless, seizures, alterations in the state of consciousness, and focal
neurological deficits also characterize HSV-2 encephalitis. HSV-2 and CMV encephalitis can coexist in patients with AIDS. The clinical features
of HSV-2 encephalitis in immunocompetent adults are generally similar to those seen with HSV-1 encephalitis, although some immunocompetent
patients with HSV-2 encephalitis have less-aggressive forms of encephalitis.
HSV Neuropathy (Zosteriform Eruption)
HSV-2 can produce pain and dermatomal distribution rash (zosteriform eruption). Herpes lesions above the neck typically result from
reactivation of HSV-1, whereas lesions below the waist result from reactivation of HSV-2, although exceptions to this rule occur. There is usually
a prodrome of diffuse neuralgia, often with malaise and fever, followed within days by the appearance of vesicles.[29] Most reports describe
lesions in one or more of the facial areas served by the trigeminal nerve, and also on the trunk, extremities and genitalia. Recurrent sciatica has](https://image.slidesharecdn.com/caseherp2-100117053949-phpapp02/75/Case-record-Herpes-simplex-type-1-encephalitis-6-2048.jpg)
![been associated with HSV.[30] A first episode can be confused with herpes zoster, but recurrent episodes of dermatomal neuralgic pain and
zosteriform eruptions are usually caused by HSV-2.[25] Although HSV neuropathy is now well documented, the exact type of HSV responsible
for each form of neuropathy is still unknown. Future DNA analysis of herpesvirus isolates, by PCR with virus-specific primers, should clarify
whether neuropathy is caused primarily by HSV-1 or HSV-2.
HSV Meningitis, Myelitis and Brainstem Encephalitis
HSV meningitis is usually caused by HSV-2. There have been no controlled trials of antiviral therapy for either isolated or recurrent HSV
meningitis, although noncontrolled experience indicates that treatment with aciclovir or related antiviral drugs might reduce the duration and
severity of attacks. Prophylactic antiviral treatment might also reduce the frequency and severity of recurrent meningitis, although its
therapeutic efficacy has not been clearly established.
HSV-2 can also cause monophasic or recurrent brainstem encephalitis and myelitis. As almost all published studies are single-case reports, it is
not possible to evaluate the efficacy of antiviral therapy for these conditions. The severity of these infections has, however, highlighted the need
for treatments, usually the same as those for HSV encephalitis. In patients with recurrent disease, valaciclovir or aciclovir have been used to
reduce the likelihood of recurrences.
Pathogenesis and Latency
HSV-2 has been isolated from normal human sacral ganglia,[28] but the physical state of HSV-2 DNA and HSV-2 gene expression in latently
infected human ganglia have not been studied nearly as extensively as the characteristics of HSV-1 in trigeminal ganglia. Latent lumbosacral
ganglionic infection can be established in mice and guinea pigs by intravaginal infection, and virus can be reactivated after sciatic nerve section
(mice) or UV irradiation (guinea pigs). Whereas HSV-1 latency is found only in cranial nerve ganglia, HSV-2 becomes latent in lumbrosacral
ganglia.
Treatment
The treatment for HSV-2 encephalitis is the same as for HSV-1.
Varicella Zoster Virus (Herpes Simplex Virus 3 or HHV-3)
Clinical Features
VZV causes chickenpox (varicella), after which the virus becomes latent in cranial nerve, dorsal root and autonomic nervous system ganglia
along the entire neuraxis. Decades later, a declining VZV-specific host immunity[31] leads to virus reactivation, usually resulting in herpes zoster
(also known as shingles), which is characterized by pain and rash restricted to 1–3 dermatomes. The NIH Shingles Prevention Study estimates
that herpes zoster affects between 600,000 and 1,000,000 Americans each year.[32] The incidence and severity of herpes zoster in
immunodeficient individuals can be viewed as a continuum, ranging from a natural decline in VZV-specific immunity with age, to more serious
host immune deficits such as those encountered in transplant recipients, or in patients with cancer or AIDS.
Because VZV is latent in most ganglia, herpes zoster can occur anywhere on the body. The most common sites are thoracic, and in the cutaneous
distribution of the ophthalmic branch of the trigeminal nerve. VZV reactivation from the geniculate (seventh cranial nerve) ganglion can cause
the Ramsay Hunt syndrome, features of which include peripheral facial weakness and rash around the ear (zoster oticus).
In many herpes zoster patients over the age of 60 years, pain—so-called postherpetic neuralgia (PHN)—continues for months or years. Although
the cause of PHN is uncertain, analyses of blood, CSF and postmortem ganglia indicate that it is related to persistent or low-grade productive
infection in ganglia.[33]
After herpes zoster, VZV can also spread to blood vessels of the brain, producing a unifocal or multifocal vasculopathy, particularly in
immunocompromised individuals. Virological analyses of patients who died from VZV vasculopathy have revealed both VZV DNA and VZV
antigen in cerebral arteries,[34] indicating active virus infection. Unifocal vasculopathy, formerly called granulomatous arteritis, is the
predominant form in elderly immunocompetent adults, and is characterized by an acute focal deficit that develops weeks to months after
contralateral trigeminal distribution herpes zoster. Multifocal vasculopathy develops in immunocompromised individuals, and is associated with
headache, fever, mental status changes and focal deficit. CSF mononuclear pleocytosis is usually present in both vasculopathies. Brain MRI
scanning reveals a single large infarct in unifocal vasculopathy; in multifocal vasculopathy, ischemic and hemorrhagic infarcts are seen in both
cortical and subcortical areas (Figure 1). Cerebral angiography reveals focal arterial stenosis. Pathological changes in affected arteries include
the presence of multinucleated giant cells, Cowdry A inclusion bodies and herpesvirus particles. Virological analysis reveals VZV DNA and
antigen in affected vessels. Vasculopathy occurs without rash, can recur, and presents with transient ischemic attacks, including posterior
ischemic optic neuropathy, remote from, and months after, acute herpes zoster.[35] Detection of VZV DNA or antibody in CSF confirms a
diagnosis of VZV vasculopathy. As infection is active and often protracted, anti-VZV IgM or IgG antibody is found in CSF, with reduced
serum:CSF ratios of VZV antibody compared with total IgG or albumin.[36] Detection of antibody to VZV in CSF, even without amplifiable
VZV DNA, can be diagnostic. Recognition of the wide spectrum of VZV vasculopathy and its proclivity to recur is essential, as effective antiviral
therapy can be curative.
Pathogenesis and Latency
The dermatomal distribution of herpes zoster lesions, combined with peak VZV titers in varicella or herpes zoster vesicles, indicate that virus
that has reactivated from latency in PNS ganglia spreads to the skin by intra-axonal transportation, although the presence of VZV in multiple
organs in disseminated zoster is most consistent with hematogenous dissemination. VZV-associated viremia and lymphotropism have been
documented during varicella, herpes zoster, PHN and zoster sine herpete, and even in normal healthy adults. During varicella, T cells are
productively infected, probably contributing to the spread of the virus. VZV DNA, transcripts and protein have been detected in mononuclear
cells (MNCs) of herpes zoster and PHN patients. With one exception,[37] attempts to recover infectious VZV from MNCs of immunocompetent
herpes zoster patients have failed.[38,39] In herpes zoster and PHN patients, lack of detectable infectious virus might be attributable to the cell-
associated nature of the virus, low-level or abortive infection of MNCs, or a decrease in the frequency of MNCs that support productive
infection, as compared with primary infection.
The detection of VZV DNA[40,41] and VZV-specific proteins[42] in blood MNCs of some patients with PHN and zoster sine herpete,[43,44,45] as
well as in tissue of patients with chronic VZV ganglionitis,[46] might reflect low-level productive infection in ganglia—a hypothesis that is
supported by favorable clinical responses in some PHN patients treated with antiviral agents.[47,48] MNCs, and in particular antigen-presenting](https://image.slidesharecdn.com/caseherp2-100117053949-phpapp02/75/Case-record-Herpes-simplex-type-1-encephalitis-7-2048.jpg)
![cells, might acquire virus while trafficking through ganglia. Ultimately, digestion of virus would account for the detection of some, but not all,
regions of the VZV genome in circulating MNCs,[49] compared with the presence of the entire virus genome in latently infected ganglia.
Digestion of viral DNA by MNCs also helps to explain why VZV DNA is found in MNCs from only 20% of patients with PHN; there would only
be a limited probability that any given fragment of viral DNA would be amplified by PCR.
Unlike HSV-1, VZV cannot be isolated from latently infected human ganglia, although nucleic acid hybridization and PCR have detected VZV
DNA in ganglia during latency.[11,50] Whereas HSV-1 latency is mostly restricted to cranial nerve ganglia, VZV is latent in cranial nerve, dorsal
root and autonomic nervous system ganglia in over 90% of healthy adults. As in the case of HSV-1, neurons are the exclusive site of latent VZV.
[51,52] Analysis of trigeminal ganglia revealed no difference in VZV copy number between the left and right trigeminal ganglia of the same
individual, but showed a wide variation of 19–3,145 copies per latently infected ganglion between individuals,[14] probably reflecting the uneven
amount of VZV encountered during primary infection. Like HSV-1, VZV DNA exists as an endless (either circular or concatameric) episome.
[53] Five VZV genes corresponding to open reading frames 21, 29, 62, 63 and 66 are transcribed, and proteins from three of these (62, 63 and 66)
have been found in latently infected ganglionic neurons. Important differences between the clinical features of HSV and VZV reactivation are
cited in Table 2 .
Treatment
As the neurological complications of VZV infection are generally caused by replication of VZV in the nervous system, inhibition of replication is
an obvious target for treatment.
Herpes Zoster
Antiviral therapy is recommended for immunocompetent adults over 50 years of age with herpes zoster, particularly if it can be initiated within
72 hours of lesion onset. The improved pharmacokinetic profiles and simpler dosing regimens of oral valaciclovir (1,000 mg three times daily for
7 days) or famciclovir (250 mg or 500 mg three times daily) have led to their use instead of aciclovir (800 mg five times daily) as the preferred
treatment. Steroids might reduce the inflammation that contributes to acute pain, provided that there are no contraindications and the potential
risk of excessive adverse effects is explained. Although they do not prevent PHN, steroids reduce acute symptoms and might facilitate a return to
normal quality of life. There is no indication for the use of topical antiviral agents to treat cutaneous herpes zoster.
The premise that the pain experienced by people with herpes zoster is primarily attributable to inflammation and necrosis of neurons led to the
suggestion that, by reducing inflammation, corticosteroids would reduce pain. The debate on the role of corticosteroid therapy in herpes zoster
has largely been resolved by two large prospective clinical trials, which demonstrated the benefit of corticosteroids in reducing the duration of
acute pain, although neither study showed any reduction in the incidence or duration of PHN among corticosteroid recipients.[54,55]
Postherpetic Neuralgia
PHN is operationally defined as pain that persists for more than 3 months after herpes zoster. Tricyclic antidepressants, such as amitriptyline or
nortriptyline (initiated at 10–25 mg and increasing weekly by 25 mg [10 mg if the patient is >65 years old or frail]), relieve pain in some patients.
Gabapentin, 300–900 mg/day up to 3,600 mg/day (provided that there is unimpaired renal function), is also recommended. Pregabalin is another
FDA-approved drug that is being used increasingly to treat PHN. A topical local anesthetic (e.g. lidocaine patch 5%) might also help.
Vasculopathy and Myelitis
Vasculopathy and myelitis are treated with intravenous aciclovir (10–15 mg/kg every 8 hours for 10–14 days). The role of antiviral therapy in
individuals presenting with rarer complications of varicella, such as cerebellar ataxia, has not been studied in a prospective or controlled
fashion; however, administration of intravenous aciclovir to such patients is likely to be appropriate. Immunocompromised patients might
require longer periods of treatment. Treatment should be discontinued if both VZV DNA and anti-VZV antibody are absent from CSF collected
at the time of treatment initiation.
Epstein–Barr Virus (Herpes Simplex Virus 4 or HHV-4)
Clinical Features
EBV causes aseptic meningitis, encephalomyeloneuritis and neuritis. The EBV neuropathies present with ophthalmoplegia, lumbosacral
plexopathy, and sensory or autonomic neuropathy. Historically, the association of virus with neurological disease was suggested by a positive
serum heterophile antibody titer, and later by the presence of EBV DNA, antibody or both in CSF.[56] EBV is latent in B cells, and caution
should be used in attributing neurological disease to EBV purely on the basis of a positive CSF PCR; in previously EBV-infected individuals,
PCR might detect EBV DNA latent in B cells that are part of the inflammatory response induced by another agent. The presence of anti-EBV
IgM or IgG antibody in CSF is more likely to be significant, particularly if there is evidence of intrathecal synthesis of EBV antibody (see below).
In a comprehensive clinical, radiological and virological analysis of four patients with EBV myeloradiculitis, encephalomyeloradiculitis and
meningoencephalomyeloradiculitis,[57] the CSF contained a mononuclear pleocytosis with elevated protein and normal glucose; in two patients,
MRI scans revealed an increased signal in the spinal cord and lumbosacral roots, but no brain swelling or focal changes. In all four patients,
EBV DNA was found in the CSF and there were reduced serum:CSF ratios of antibody to EBV, but not to total IgG or albumin, consistent with
intrathecal antibody synthesis. Residual neurological deficits were evident. Disappearance of viral DNA from CSF at the time of improvement of
neurological disease, particularly before a decline in CSF white blood cells, supports the theory that CNS disease results from direct invasion of
the CNS by virus. EBV DNA has been found in brain biopsies from affected patients.[58] CSF from patients with EBV infection of the nervous
system contains mainly EBV-specific CD8+ T cells, a few CD4+ T cells, and no CD19+ B cells.[59] CD8+ cells from one patient with EBV
infection were shown to contain EBV DNA, and had unrestricted cytotoxic activity.[60] Patients with postinfectious complications of EBV
infection have a low EBV load and a high CSF leukocyte count. Encephalitis is characterized by intense viral replication and vigorous
inflammation, whereas CNS lymphoma is associated with a limited inflammatory response.
EBV-associated CNS lymphomas are rare in immunocompetent individuals. EBV triggers AIDS-associated primary CNS lymphoma (PCNSL)
and post-transplantation lymphoproliferative disorder (PTLD). EBV DNA can be found in tumor tissue in both of these conditions[61] and in
CSF of patients with AIDS-associated PCNSL. Deficiency in cytotoxic T lymphocytes seems to permit the outgrowth of EBV-transformed B cells,
resulting in PTLD. Administration of autologous EBV-specific cytotoxic T cells has been shown to produce specific killing of EBV-transformed B
cells in patients with PTLD.[62]
Pathogenesis and Latency](https://image.slidesharecdn.com/caseherp2-100117053949-phpapp02/75/Case-record-Herpes-simplex-type-1-encephalitis-8-2048.jpg)
![EBV infects B and T lymphocytes of more than 90% of the general population before adulthood. Primary infection results in transient viremia,
followed by a rapid immune response. EBV replicates in the oropharynx and is transmitted through oral secretions. Primary EBV infection
frequently results in infectious mononucleosis. EBV is also associated with nasopharyngeal carcinoma, Burkitt's lymphoma, Hodgkin's disease,
and lymphoproliferative disease in immunocompromised individuals.[63] It is estimated that neurological complications occur in 1–5% of
individuals with infectious mononucleosis. Given the widespread prevalence of EBV infection, the burden of neurological disease is probably
underestimated; for example, virtually all CNS lymphomas in AIDS patients contain EBV DNA.[61]
EBV establishes latency in B lymphocytes, which adopt restricted patterns of EBV gene expression. EBV latency has been classified into three
types: type I (Burkitt's lymphoma), in which only the EBV-encoded nuclear antigen (EBNA-1) is expressed; type II (nasopharyngeal carcinoma),
in which EBNA-1 is coexpressed along with the latent membrane proteins LMP-1, LMP-2A and LMP-2B; and type III (lymphoproliferative
diseases in immunosuppressed individuals), in which five EBNAs and the three LMPs are expressed. Autoactivation of gene expression by LMP-
1 might be important in type II latency, and EBNA-2-dependent regulation of LMP-1 expression appears to be important in type III latency.[64]
The precise mechanism by which EBV reactivates remains unknown, however.
Treatment
There is no known effective antiviral or other accepted treatment for EBV.
Cytomegalovirus (Herpes Simplex Virus 5 or HHV-5)
Clinical Features
CMV produces neurological disease predominantly in infants with congenital infection. Although most congenital CMV infections are
asymptomatic, many carriers develop sensorineural hearing loss and intellectual handicaps. Other neurological complications include
microcephaly, seizures, hypotonia and spasticity. In immunocompetent adults, the most common neurological complication of CMV infection is
Guillain–Barré syndrome, and CMV has also been associated with acute brachial plexopathy.[65]
The natural history of CMV infection of the CNS in immunocompetent adults is unknown. CMV encephalitis was first described in renal[66]
and bone marrow transplant recipients, and CMV infections in patients with AIDS can cause encephalitis, myelitis, retinitis and polyradiculitis.
The CSF might show neutrophilic or mononuclear pleocytosis, or an elevated protein and depressed glucose content, and MRI can reveal
enhancement in the ventricular ependyma. Focal disease has been attributed to CMV vasculitis or demyelination. Characteristic owl-eyed
cytomegalic inclusions and CMV-specific antigens have been found in brain tissue and blood vessels of AIDS patients with subacute
encephalopathy. Complicating the picture, however, is the additional presence of HSV-2. It is difficult, therefore, to attribute specific symptoms
and signs to CMV.
CMV polyradiculopathy in patients with AIDS begins insidiously as a cauda equina syndrome with paresthesias and distal weakness (usually
asymmetric), incontinence, and sacral-distribution sensory loss. CSF pleocytosis is usually present, often with a predominance of
polymorphonuclear leukocytes. Most cases have elevated CSF protein and hypoglycorrhachia. CMV can be isolated from CSF, and CMV DNA
can be amplified by CSF PCR. Postmortem examination reveals inflammation, necrosis and focal vasculitis of nerve roots, along with typical
CMV inclusions.[67]
Pathogenesis and Latency
CMV has a genome nearly twice as large as those of other HHVs, one which encodes more than 200 genes. During primary infection, CMV
replicates in leukocytes and vascular endothelial cells, after which virus becomes latent principally in bone marrow progenitor cells and myeloid
cells.[68] Human CMVs show DNA sequence variability[69] and, when adapted to in vitro growth, acquire extensive DNA deletions.[70]
Reactivation and shedding of infectious human CMV follows immunosuppression, differentiation of latently infected progenitor cells, or
allogeneic organ transplant.[71] After reactivation, CMV eludes immunological clearance by suppressing MHC class I and class II responses,
inhibiting apoptotic cell death, and sequestering chemokines.[72]
Treatment
There have been no randomized controlled studies of antiviral therapy in adults with CMV infection of the nervous system. An international
panel that developed guidelines for treatment of CMV diseases in adults with AIDS receiving highly active antiretroviral therapy[73]
recommended treatment with intravenous ganciclovir, intravenous foscarnet, or a combination of the two drugs. Patients who develop CMV
polyradiculopathy while on ganciclovir or foscarnet therapy should receive combination therapy with both drugs. Patients not on treatment at
the time of disease onset could be treated with either drug as monotherapy, and switched to combination therapy if they progressed clinically
and/or had persisting CSF pleocytosis (a potential marker of drug failure). Reports of antiviral therapy for CMV-associated mononeuritis
multiplex or painful symmetrical neuropathy are too limited to permit conclusions about efficacy of antiviral therapy.
Human Herpesvirus 6
Clinical Features
HHV-6 causes roseola, a common childhood exanthematous disease. Neurological complications can result from direct invasion of the CNS.
HHV-6 DNA has been detected in the CSF of patients with febrile seizures[74] and in the brains of patients with fulminant hepatitis.[75] HHV-6
antigen was found in brain tissue from a fatal case of roseola.[76] HHV-6 causes encephalitis primarily after bone marrow or stem cell
transplantation,[77] and might present as limbic encephalitis.[78] Diagnosis is confirmed by detection of HHV-6 DNA in the CSF. On the basis of
the presence of HHV-6 DNA and increased levels of antibody to HHV-6 in blood and CSF of patients with multiple sclerosis (MS), HHV-6 has
been implicated in the pathogenesis of the disease. HHV-6 is, however, found in only a minority of MS patients, and HHV-6 DNA and elevated
HHV-6 antibody levels are also seen in patients with other neurological diseases.
Human herpesvirus-6 infection, febrile seizures, and encephalitis
Another recent important development has been the identification of human herpesvirus-6 (HHV-6) and the elucidation of its role in human
disease. HHV-6, a member of the Herpesviridae family of viruses, was discovered in 1986. Although biologically similar to cytomegalovirus
(CMV), the virus was recognized as serologically and genetically distinct from other human herpes viruses [89]. Soon thereafter, HHV-6 was
noted to be ubiquitous and a principal cause of the common childhood disease exanthem subitum (also referred to as roseola or sixth disease)
[89]. From the neurologic perspective, HHV-6 is of importance because of its linkage to febrile seizures and encephalitis.](https://image.slidesharecdn.com/caseherp2-100117053949-phpapp02/75/Case-record-Herpes-simplex-type-1-encephalitis-9-2048.jpg)
![Exanthem subitum typically occurs between 6 months and 2 years of age and is characterized by an abrupt rise in temperature, often to
approximately 40°C, followed several days later by a rapid defervescence that coincides with the emergence of an erythematous maculopapular
rash that can persist for several days. Fever is a prominent component of the disease, and it has been recognized for many years that febrile
seizures occur commonly during the early (febrile) stage of exanthem subitum.
The linkage of exanthem subitum to febrile seizures, along with the identification of HHV-6 as the causative pathogen for exanthem subitum, led
naturally to the hypothesis that HHV-6 infection may underlie many cases of febrile seizures. This has proven to be true. Multiple studies have
demonstrated that a substantial proportion of febrile seizures occur in children with a primary HHV-6 infection [89]. The risk of febrile seizures
is high during primary infection with HHV-6, whether or not the child develops the rash of exanthem subitum.
In addition to increasing the risk of febrile seizures, a primary infection with HHV-6 may increase the severity of the febrile convulsion. Partial
seizures, prolonged seizures, and repeated seizures are more common when the febrile seizures are associated with HHV-6 infection than when
the seizures are caused by other pathogens [89]. Furthermore, reports have suggested that HHV-6 may persist within the CNS [89] and that
reactivation of HHV-6 may be associated with recurrent febrile seizures [89]. This remains controversial, however, and at least one study has
found that children with an initial febrile seizure induced by HHV-6 infection have a lower risk of febrile seizure recurrence than do children
whose febrile seizures were triggered by other pathogens [89].
Although the association of HHV-6 infection with febrile seizures is well documented, the mechanism underlying this association remains
unclear. One possibility is that HHV-6 directly invades brain parenchyma during a primary infection and that the febrile convulsion is triggered
by brain infection. This hypothesis is supported by the findings that HHV-6 is a neurotropic virus that can replicate in glial cells and in brain
endothelial cells [89]. Identification of HHV-6 DNA in CSF of patients with febrile seizures further supports the notion that the brain is invaded
during HHV-6 infection [89]. Other studies have not found HHV-6 DNA in the CSF of patients with febrile seizures [89], however, and the
question of brain invasion in typical cases of HHV-6 infection remains controversial.
A second possibility is that the seizures are triggered not by direct viral invasion of the CNS but by the production of a toxin or inflammatory
molecule liberated as part of the viral infection. Cytokines, for example, are produced at high levels by circulating monocytes during HHV-6
infection and can act on neurons to lower the seizure threshold [89].
A third possibility is that the febrile convulsions are not directly related to the HHV-6 infection per se but to a characteristic of the fever induced
by the infection. Febrile seizures occur almost exclusively in children, presumably because the immature brain has a lower seizure threshold to
the destabilizing effects of temperature elevation. A hallmark of exanthem subitum is a rapid onset of high fever. Thus, children infected with
HHV-6 may be at greater risk for seizures than children with other infections because of the rapidity of high fever onset with HHV-6. Arguing
against this possibility, however, are the findings that neither the maximal temperature nor the duration of fever before the seizure differed
between febrile children who developed seizures and those who did not [2,16].
In a few patients, HHV-6 produces encephalitis. Encephalitis caused by HHV-6 has been observed both in immunocompromised patients and in
children whose immune function was apparently normal [89]. As might have been anticipated from the association of HHV-6 with convulsions,
seizures, and status epilepticus are common presenting signs of HHV-6 encephalitis [89]. Confusion and headache, common symptoms of
encephalitis in general, are also commonly observed in encephalitis caused by HHV-6. When a patient develops encephalitis in association with
exanthem subitum, the signs of encephalitis usually arise during the febrile phase of the illness before the onset of rash [89].
Whether invasion of the brain by HHV-6 is necessary to produce encephalitis is unresolved. In some cases of HHV-6 encephalitis, viral antigen
has been detected within brain astrocytes and neurons [89], the location of viral antigen has been correlated with the sites of neuropathologic
changes [19], and HHV-6 DNA has been detected in CSF. These findings strongly suggest that viral invasion occurs and that the presence HHV-6
within the brain plays a key role in the pathophysiology. Nevertheless, other studies have noted the distinct absence of viral antigen from the
brain [21] and of viral DNA from the CSF [19] in cases of HHV-6 encephalitis. These results suggest that in at least some cases of HHV-6
encephalitis, the brain dysfunction may be caused by an extrainfectious process.
Most children who develop HHV-6 encephalitis have a good outcome with no neurologic sequelae. Some children are left with serious permanent
neurologic problems, however, including mental retardation, hemiplegia, and epilepsy [89]. Occasional cases of HHV-6 encephalitis have been
fatal [89].
Figure 9. Schematic representation of HHV-6 and HHV-7 genomes. The genomes are colinear. Homologies are 46.6% to 84.9%. Red blocks
represent the herpesvirus core genes, numbered from I to VII. Yellow blocks represent ß-herpesvirus subfamily-specific genes (from U2 to U14).
Green blocks indicate genes present only in the Roseolovirus genus, i.e., in HHV-6 and HHV-7. Only three ORFs (U22, U83 and U94) are present
in HHV-6 and absent from HHV-7
Pathogenesis and Latency
As HHV-6 becomes latent in cells of the monocyte–macrophage lineage,[79] the detection of HHV-6 DNA and antigen in brain tissue is likely to
reflect HHV-6 reactivation from latency in blood MNCs trafficking through the brain in patients with inflammatory CNS disease. CD46 is an
HHV-6 receptor that is expressed mostly in macrophages and cells lining blood vessels, and less often in cells of neuronal origin. HHV-6
expresses proinflammatory cytokines that are cytotoxic to oligodendrocytes and induce caspase-independent apoptosis.[80] HHV-6 has also been
shown to inhibit G1–S-phase transition in glial precursor cells, which are important in remyelination.[81] Reactivation of latent HHV-6 is
frequently associated with drug-induced hypersensitivity syndrome, possibly because of the acute decrease in serum IgG levels observed in these
patients.
Treatment](https://image.slidesharecdn.com/caseherp2-100117053949-phpapp02/75/Case-record-Herpes-simplex-type-1-encephalitis-10-2048.jpg)
![There have been no randomized controlled trials of antiviral therapies in either immunocompetent or immunocompromised patients.
Ganciclovir and foscarnet have been used, but their efficacy is unknown. The outcome in immunocompetent patients does not appear to be
substantially better than that in immunocompromised individuals. Real-time fluorescent probe CSF PCR has been used to follow the effects of
antiviral therapy on HHV-6 DNA levels in the CSF of 11 hematopoietic stem cell recipients with HHV-6 infection (8 of whom had HHV-6
encephalitis).[82] All patients were treated with ganciclovir, foscarnet or both. Within 3 weeks of commencing antiviral therapy, median CSF
viral load had decreased from 25,000 copies/ml to 100 copies/ml, although the difference was not statistically significant (P = 0.13). Four of the
eight patients with HHV-6 encephalitis improved clinically, with viral load declining concurrently with antiviral therapy.
Human Herpesvirus 7
HHV-7 is closely related to HHV-6, and can induce reactivation of the latter in vitro. HHV-7 was first isolated from CD4+ T cells of a healthy 26-
year-old under conditions that promote T-cell activation.[83] HHV-7 is ubiquitous and is acquired in childhood. There is a single report of
primary HHV-7 encephalitis in an immunocompetent adult, in whom HHV-7 DNA was found in the CSF,[84] and another report of meningitis
and optic neuritis resulting from reactivation of HHV-7 in a stem cell transplant recipient.[85] HHV-7 infection in children has been linked with
seizures and encephalitis through the detection of intrathecal synthesis of antibody against the virus.[86]
Kaposi's Sarcoma-associated Herpesvirus (Human Herpesvirus 8)
Kaposi's sarcoma-associated herpesvirus (KSHV or HHV-8) is the most recently discovered HHV. Using PCR-based subtractive analysis, Chang
et al.[87] detected segments of the KSHV genome in tumor cells of Kaposi's sarcomas, the most common tumors seen in patients with AIDS.
KSHV is also associated with primary effusion lymphoma and specific forms of multicentric Castleman disease, both of which are B-cell
lymphoproliferative disorders. KSHV becomes latent in B cells. The viral genome is episomal, and its complete sequence (137 kilobase pairs) has
been determined. Like EBV, KSHV encodes a latency-associated nuclear antigen, which tethers the virus episome to the host chromosome. This
tethering not only ensures that virus DNA replication is synchronous with host chromosome replication, but also represses activation of virus
genes associated with replication. Expression of a lytic HHV-8 gene that encodes a viral G-protein-coupled receptor can lead to the formation of
Kaposi's sarcoma-like lesions in mice.[88] To date, no neurological disease has been associated with KSHV infection.
Table 3. Management of herpes infection
Disease Pharmacological Therapy
Herpes Simplex Virus 1 Intravenous aciclovir (10 mg/kg body weight three times a day for 14–21 days) is the standard
treatment for HSV-1 encephalitis in adults. Steroids are sometimes given as well, although data
regarding the efficacy of this approach remain anecdotal. Cognitive impairment and seizures are
significant neurological sequelae in treated patients.
Herpes Simplex Virus 2 Intravenous aciclovir (10 mg/kg body weight three times a day for 14–21 days) is the standard
treatment for HSV-1 encephalitis in adults. Steroids are sometimes given as well, although data
regarding the efficacy of this approach remain anecdotal. Cognitive impairment and seizures are
significant neurological sequelae in treated patients.
Varicella Zoster Virus (Herpes Simplex Intravenous aciclovir (10 mg/kg body weight three times a day for 14–21 days) is the standard
Virus 3) treatment for HSV-1 encephalitis in adults. Steroids are sometimes given as well, although data
regarding the efficacy of this approach remain anecdotal. Cognitive impairment and seizures are
significant neurological sequelae in treated patients.
Epstein–Barr Virus (Herpes Simplex There is no known effective antiviral or other accepted treatment for EBV.
Virus 4)
Cytomegaloviru (Herpes Simplex Virus There have been no randomized controlled studies of antiviral therapy in adults with CMV infection
5) of the nervous system. An international panel that developed guidelines for treatment of CMV
diseases in adults with AIDS receiving highly active antiretroviral therapy[73] recommended
treatment with intravenous ganciclovir, intravenous foscarnet, or a combination of the two drugs.
Patients who develop CMV polyradiculopathy while on ganciclovir or foscarnet therapy should
receive combination therapy with both drugs. Patients not on treatment at the time of disease onset
could be treated with either drug as monotherapy, and switched to combination therapy if they
progressed clinically and/or had persisting CSF pleocytosis (a potential marker of drug failure).
Reports of antiviral therapy for CMV-associated mononeuritis multiplex or painful symmetrical
neuropathy are too limited to permit conclusions about efficacy of antiviral therapy.
Herpes Simplex Virus 6 There have been no randomized controlled trials of antiviral therapies in either immunocompetent or
immunocompromised patients. Ganciclovir and foscarnet have been used, but their efficacy is
unknown. The outcome in immunocompetent patients does not appear to be substantially better than
that in immunocompromised individuals. Real-time fluorescent probe CSF PCR has been used to
follow the effects of antiviral therapy on HHV-6 DNA levels in the CSF of 11 hematopoietic stem cell
recipients with HHV-6 infection (8 of whom had HHV-6 encephalitis).[82] All patients were treated
with ganciclovir, foscarnet or both. Within 3 weeks of commencing antiviral therapy, median CSF
viral load had decreased from 25,000 copies/ml to 100 copies/ml, although the difference was not
statistically significant (P = 0.13). Four of the eight patients with HHV-6 encephalitis improved
clinically, with viral load declining concurrently with antiviral therapy.
Herpes Simplex Virus 7 NA
Kaposi's Sarcoma-associated NA
Herpesvirus (Herpes Simplex Virus 8)
SUMMARY](https://image.slidesharecdn.com/caseherp2-100117053949-phpapp02/75/Case-record-Herpes-simplex-type-1-encephalitis-11-2048.jpg)
![SUMMARY
Most HHVs can cause serious neurological disease of the PNS and CNS through primary infection or following virus reactivation from latently
infected human ganglia or lymphoid tissue. The neurological complications include meningitis, encephalitis, myelitis, vasculopathy, acute and
chronic radiculoneuritis, and various inflammatory diseases of the eye. Disease can be monophasic, recurrent or chronic. Recognition of the
clinical patterns and imaging characteristics of disease produced by different herpesviruses is important, because infection by many of the
herpesviruses can be treated successfully. Early diagnosis and proper treatment are essential to a favorable outcome. Virological confirmation of
neurological disease relies on the detection of herpesvirus-specific DNA in bodily fluids or tissues, herpesvirus-specific IgM in blood, or
herpesvirus-specific IgM or IgG antibody in cerebrospinal fluid. HSV-1, HSV-2, VZV and CMV are the most treatable herpesviruses.
Table 4. The neurology of herpes viruses infection
Virus Comment
Herpes simplex virus 1 Herpes simplex virus 1 (HSV-1) encephalitis predominantly involves the orbital surface of the frontal lobes and
medial surface of the temporal lobes, resulting in areas of increased T2 signal on MRI
Herpes simplex virus 2 Herpes simplex virus 2 (HSV-2) is the primary cause of recurrent meningitis
Varicella zoster virus After varicella, the varicella zoster virus (VZV) becomes latent in ganglia along the entire neuraxis; its reactivation
(Human Herpesvirus 3) can lead to herpes zoster, vasculopathy, myelitis, necrotizing retinitis or zoster sine herpete
Epstein–Barr virus The neurological complications of Epstein–Barr virus are diverse, and include meningitis, encephalitis, myelitis,
(Human Herpesvirus 4) radiculoneuropathy, and even autonomic neuropathy
cytomegalovirus (CMV) The most common neurological complication of cytomegalovirus (CMV) is poly-radiculoneuropathy in
(Human Herpesvirus 5) immunocompromised individuals
Human Herpesvirus 6 HHV-6 causes roseola, a common childhood exanthematous disease. Neurological complications can result from
direct invasion of the CNS. HHV-6 DNA has been detected in the CSF of patients with febrile seizures[74] and in the
brains of patients with fulminant hepatitis.[75] HHV-6 antigen was found in brain tissue from a fatal case of roseola.
[76] HHV-6 causes encephalitis primarily after bone marrow or stem cell transplantation,[77] and might present as
limbic encephalitis.[78]
Human Herpesvirus 7 HHV-7 is closely related to HHV-6, and can induce reactivation of the latter in vitro. HHV-7 was first isolated from
CD4+ T cells of a healthy 26-year-old under conditions that promote T-cell activation.[83] HHV-7 is ubiquitous and
is acquired in childhood. There is a single report of primary HHV-7 encephalitis in an immunocompetent adult, in
whom HHV-7 DNA was found in the CSF,[84
Human Herpesvirus 8 Kaposi's sarcoma-associated herpesvirus (KSHV or HHV-8) is the most recently discovered HHV. Using PCR-based
subtractive analysis, Chang et al.[87] detected segments of the KSHV genome in tumor cells of Kaposi's sarcomas,
the most common tumors seen in patients with AIDS.
Addendum
A new version of this PDF file (with a new case) is uploaded in my web site every week (every Saturday and remains available till Friday.)
To download the current version follow the link "http://pdf.yassermetwally.com/case.pdf".
You can also download the current version from my web site at "http://yassermetwally.com".
To download the software version of the publication (crow.exe) follow the link: http://neurology.yassermetwally.com/crow.zip
The case is also presented as a short case in PDF format, to download the short case follow the link: http://pdf.yassermetwally.com/short.pdf
At the end of each year, all the publications are compiled on a single CD-ROM, please contact the author to know more details.
Screen resolution is better set at 1024*768 pixel screen area for optimum display.
For an archive of the previously reported cases go to www.yassermetwally.net, then under pages in the right panel, scroll down and click on
the text entry "downloadable case records in PDF format"
Also to view a list of the previously published case records follow the following link (http://wordpress.com/tag/case-record/) or click on it if
it appears as a link in your PDF reader
REFERENCES
References
1.Lakeman FD and Whitley RJ (1995) Diagnosis of herpes simplex encephalitis: application of polymerase chain reaction to cerebrospinal fluid
from brain-biopsied patients and correlation with disease. National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study
Group. J Infect Dis 171: 857-863
2.Weil AA et al. (2002) Patients with suspected herpes simplex encephalitis: rethinking an initial negative polymerase chain reaction result. Clin
Infect Dis 34: 1154-1157
3.Westmoreland BF (1987) The EEG in cerebral inflammatory processes. In Electroencephalography: Basic Principles, Clinical Applications,
and Related Fields, edn 2, 259-273 (Eds Niedermeyer E and Lopes da Silva FH) Baltimore-Munich: Urban and Schwarzenberg
4.Smith JB et al. (1975) A distinctive clinical EEG profile in herpes simplex encephalitis. Mayo Clin Proc 50: 469-474
5.Zimmerman RD et al. (1980) CT in the early diagnosis of herpes simplex encephalitis. Am J Radiol 134: 61-66
6.Schroth G et al. (1987) Early diagnosis of herpes simplex encephalitis by MRI. Neurology 37: 179-183](https://image.slidesharecdn.com/caseherp2-100117053949-phpapp02/75/Case-record-Herpes-simplex-type-1-encephalitis-12-2048.jpg)




This document presents a detailed case study of a 40-year-old diabetic woman with herpes simplex virus type 1 (HSV-1) encephalitis, highlighting her clinical presentation, radiological findings, and diagnostic tests, including cerebrospinal fluid analysis and electroencephalography. It discusses the pathogenesis of HSV-1, its ability to establish latency, and the typical neurological complications associated with it. The document concludes with treatment recommendations, primarily emphasizing intravenous acyclovir as the standard therapy for managing HSV-1 encephalitis and the importance of timely intervention.























































































